


ラウリル硫酸をドープしたポリアニリンとポリスチレンの新しいナノコンポジット
要約
この作業は、ポリスチレン(PS)とドープされたポリアニリン(PANI)の新しいコアシェルナノコンポジットの合成と調査に重点を置いています。 15〜30 nmのサイズのPSナノ粒子を含むラテックスは、水媒体中でのスチレンのマイクロ乳化重合によって調製されました。 PS / PANIナノコンポジットは、ドーパントと可塑剤の両方として機能するラウリル硫酸(LSA)の存在下で、PSラテックス媒体中でアニリンを化学酸化重合することによって合成されました。合成されたナノコンポジット中のPANIの実際の含有量は、UV-Vis分光法によって決定されました。ナノコンポジットの組成とドープされたポリアニリンの酸化状態は、FTIR分光法によって特徴づけられました。ナノコンポジットナノ粒子のコアシェル形態は、透過型および走査型電子顕微鏡によって証明されました。これらのナノコンポジットの空気中の導電率と熱的挙動は、ドープされたポリアニリン含有量に非線形に依存するだけでなく、酸ドーパントの可塑化特性とポリアニリンシェルの存在の両方によって強く影響されることがわかりました。これらのナノコンポジットをセンサー材料として利用できる可能性が実証されています。
背景
ポリアニリン(PANI)は、独自の物理的および化学的特性、高い安定性、低価格などを備えていることはよく知られています。これにより、マイクロエレクトロニクスやオプトエレクトロニクス、センサー、エレクトロクロミックデバイスなどのさまざまなハイテク分野での多機能アプリケーションが可能になります。 、バッテリー、スーパーキャパシターなど[1、2]。 PANIの加工性と適用性は、さまざまな物品に容易に形成できる可溶性または溶融性の一般的なポリマーとの複合材料またはナノ複合材料で使用すると、大幅に改善できます[3]。そのような材料の様々な調製方法の中で、水ベースの酸性化ラテックスまたはナノ粒子または他のポリマーの(サブ)ミクロンサイズの粒子(異なる界面活性剤または非イオン性ポリマーで安定化)を含む分散液中でのアニリンの酸化重合は1つと見なされます最も効果的なアプローチの[3]。このアプローチにより、コアがポリマー(ナノ)粒子であり、シェルがPANIで形成されているコアシェルタイプの多機能複合材料またはナノ複合材料を得ることができます[3,4,5,6,7,8,9,10]。コアシェル形態の形成を促進するために、アニリンはその塩として重合されます。これは、添加された酸ドーパント、通常はHCl([8、10]など)とのアニリン相互作用のためにラテックス媒体に現れるか、または市販されています。アニリン塩酸塩(例、[4、7])。多くの場合、ラテックス重合媒体の安定性は、非イオン性またはイオン性安定化添加剤(より多くの場合、ドデシル硫酸ナトリウム/ラウリル硫酸ナトリウム(SDS / SLS)[3,4,5,6,7,8,9、 10])。しかし、そのような材料を準備するための代替方法が開発されました。この方法では、界面活性剤、可塑剤、および酸ドーパントの特性を統合する表面活性酸(たとえば、ドデシルベンゼンスルホン酸-DBSA)を使用するため、上記の追加のHClまたは他の酸ドーパントの使用を回避できます[11、12]。
さまざまな有機または無機の巨視的(ガラスまたは石英、ポリマーフィルム、繊維など)および微視的(ポリスチレンラテックス、シリカまたはチタニア、またはポリマー粒子など)基板の表面でのPANI層またはシェルの形成の機構的側面(テンプレート)は、主に表面での正に帯電したアニリニウムカチオンのその場吸着重合に関して多くの出版物で議論されており、通常、事前吸着/グラフト化アニオン/官能基の負電荷を帯びています[13、14、15、16、17]。 。吸着されたアニリニウムカチオンは、酸化剤開始剤の添加直後に重合することが一般的に認められている。当然、非吸着のアニリニウムカチオンも重合プロセスに関与し、正に帯電したオリゴマーおよびポリマー分子を形成します。これらの分子は同じ表面で沈殿/吸着するため、PANIシェルの厚さが増加します。
別のアプローチでは、アニリンモノマーを最初にポリスチレン(PS)ラテックスに添加し、PSコア粒子によって中性の形で3日間吸収されました[9]。酸化剤開始剤(過硫酸アンモニウム、APS)を添加した後、導電性ポリアニリン膜が粒子界面に形成され、両方の試薬を分離します。電子はポリアニリン膜を介してアニリン分子から酸化剤分子に移動するため、ポリアニリンによるラテックス粒子の古典的なコーティングで得られる上記のコアシェル形態とは対照的に、ポリアニリンはPSラテックス粒子の内部に徐々に浸透します[9]。このアプローチの別のバリエーションでは、PS粒子を中性アニリンモノマーで12時間膨潤させた後、APS、次に塩酸を反応媒体に添加しました[18]。 HClの添加により、粒子から放出されたアニリン分子がアニリニウムカチオンに変換され、APSによる化学酸化によって重合されました。この場合、形成されたPS / PANI複合材料の明確なコアシェル構造が確認されました[18]。
PSは、その優れた熱的および化学的安定性、機械的特性、生体適合性など[19]だけでなく、おそらく形状の良いナノ/サブミクロン/ミクロンの合成に便利なため、コアポリマーコンポーネントなどの材料で頻繁に使用されます。サイズの粒子は、特定の用途の(ナノ)複合材料に非常に適しています。たとえば、ドープされたPANIでコーティングされたミクロン/サブミクロンサイズのPS粒子は、静電加速器で使用され、荷電粒子を超高速[20]または電気粘性流体[21]などで加速することができました。他のコアシェル(ナノ)と同様です。複合材料[22] PS / PANIは、明らかにセンシングアプリケーションの可能性を秘めています。しかし、私たちの知る限り、PS / PANIコアシェル(ナノ)コンポジットをセンシング材料として使用することに関する情報は不足しています。それにもかかわらず、最近、トルエンに溶解したPSをカンファースルホン酸をドープしたPANI粒子と混合すると、アンモニアに敏感な複合膜の形成に適した分散液が得られることが示されました[23]。興味深いことに、これらのブレンドはガス状アンモニアに対して非常に高い応答を示しましたが、つまり(Δ R / R 0 )×100〜73%(20 ppm)、高濃度のアンモニアに対する反応はそれほど変わらず、100 ppmで最大〜90%にすぎませんでした([23]の図11)。この弱い応答濃度の振る舞いは、これらのブレンドの溶液調製により、敏感なPANIクラスターの一部のみが分析物分子に容易にアクセスでき、他の部分はPSマトリックスによってスクリーニングされ、センシング材料に拡散制限を与えることを示唆しています。したがって、スクリーニングされていないPANI表面を備えたPS / PANIコアシェル複合材料は、溶液で調製されたブレンド材料と比較して、改善された検知挙動を示す可能性があると推測できます[23]。
上記の議論に基づいて、私たちの仕事は主に、ラウリル硫酸(LSA)をドープしたPANIを用いたPSナノ粒子の新しいコアシェルナノコンポジットの合成と、それらの実用的に重要な特性(形態、化学構造、導電性、熱安定性)の調査に集中しました。 。センシング材料としてのそれらの潜在的な適用性も推定された。 LSAの選択は、主に3つの前提条件に基づくナノコンポジットの重要な機能です。(1)コアPSナノ粒子合成の段階で使用される界面活性剤SLSと酸ドーパントの両方で同じラウリル硫酸塩表面活性アニオンPANIシェル合成の段階で使用されるLSAは、(2)反応媒体を酸性化する界面活性官能化プロトン酸ドーパントとして機能し[24、25]、(3)アニリニウム塩を形成します(すなわち、表面活性反応性モノマーまたはサーフマー)ナノサイズのPANIシェルおよび構造の形成を促進します[25、26]。
メソッド
資料
アニリン(メルク)とスチレン(試薬グレード、ウクライナ)を真空下で蒸留し、アルゴン下で3〜5℃で保存しました。酸化剤の過硫酸カリウム(KPS)(ウクライナ)、陰イオン界面活性剤のラウリル硫酸ナトリウム(SLS、同義語としてドデシル硫酸ナトリウム-SDS、Aldrich)は試薬グレードであり、さらに精製することなく使用しました。ラウリル硫酸(LSA)は、KU-2-8樹脂(ウクライナ)とのイオン交換反応を介してSLSから調製されました。
PSラテックスの準備
PSナノ粒子ラテックスは、他の場所で説明されている方法[27]に従って、スチレンのラジカル重合によって調製されました。つまり、スチレンは、酸化剤開始剤KPSを含むSLSのミセル水溶液で、次のように重合されました。2gのスチレンを、0.01gのNaH 2 > PO 4 、0.2 g SLS、および0.01 g KPS、10 mlの水、70°C、アルゴン雰囲気。混合物を70℃でさらに3時間撹拌し、次に90℃でさらに1時間撹拌した。最終的な重合混合物を室温まで冷却し、MWCO 3500Daを使用したセルロース膜を蒸留水に対して48時間透析することにより精製しました。
PS / PANI-LSAナノ粒子の調製
PSラテックスでのアニリン重合は、他の場所で説明されている方法[28、29]と同様に、反応混合物成分の次の比率で実行されました。アニリン/ LSA =1 / 1.5(mol / mol)およびアニリン/酸化剤=1 / 1.25 (mol / mol)10°Сで。重合混合物中のPSナノ粒子に対するアニリンの初期重量比は、1〜10 wt%の範囲の最終的なナノコンポジット中の脱ドープされたポリアニリンの予想される理論量によって事前に決定されました。つまり、重合混合物の調製の最初の段階で、計算された量の酸をターゲットのPSラテックス部分に添加し、室温で30分間撹拌しました。第2段階では、計算された量のアニリンをこの酸性化PSラテックスに添加し、1時間攪拌して室温でアニリニウム塩を完全に形成させた後、調製した混合物を10°Cで30分間冷却しました。第3段階では、蒸留水中の10°Cに予冷したKPS溶液の計算量を反応混合物に滴下し、続いて10°Cで24時間撹拌しました。アニリンの重合が完了した後、得られたPS / PANI-LSAラテックスを、蒸留水に対するセロハン膜を介した3日間の透析によって精製しました。精製されたナノコンポジットを周囲条件で乾燥させて粉末条件を視覚的に乾燥させた後、一定の重量に達するまで60°Cで真空下で乾燥させました。参照の純粋なPANI-LSAサンプルは、PSナノ粒子の非存在下で水溶液中で同じ条件下で合成されました。
特性評価
合成されたナノコンポジットの実際のPANI含有量は、分光光度計Cary 50(Varian)を使用して、N-メチル-2-ピロリドン(NMP)中の溶液のUV-Vis分光分析によって[29]と同様に決定されました。つまり、最初の段階では、通常、乾燥ナノコンポジットを0.3 wt%のアンモニア水溶液に24時間脱ドープした後、蒸留水で洗浄し、一定の重量に達するまで60°Cで真空乾燥しました。第2段階では、脱ドープ粉末ナノコンポジットの固定部分をNMPに溶解し、NMPのアスコルビン酸溶液と混合して、ロイコエメラルジン塩基(LB、PANIの完全還元型)を得ました。第3段階では、LB濃度は、前に作成した検量線を使用して、343nmでの1mm石英キュベットでのこの溶液のUV吸収から計算されました。次に、第4段階で、このLB濃度を、ナノコンポジットの実際のデドープされたPANI含有量に対して再計算しました。次に、後者の含有量を使用して、このPS / PANI-LSAナノコンポジットにドープされたPANI-LSA含有量を推定しました。この最終的な再計算は、[28、29]と同様に、PANIのLSAとイミン窒素の理論的な化学量論比、つまりPANI:LSA =1:0.5に基づいています。 [28]に従って、この再計算を単純化および明確にするために、LSAのみを使用した完全なPANIドーピングを仮定しました。合成されたナノコンポジットの組成とその表記法を表1に示します。
<図>PS / PANI-LSAナノコンポジットのフーリエ変換赤外(FTIR)スペクトルと、KBrを含むペレットの純粋なPANI-LSAサンプルを、1 cm -1 の解像度で記録しました。 Bruker Vertex70分光計を使用。
透過型および走査型電子顕微鏡(TEMおよびSEM)画像は、それぞれJEOLJEM-1400およびHitachiS4800顕微鏡で取得しました。 TEM測定用のサンプルは、2μLのサンプル水分散液をカーボンまたはformvarでコーティングされた200メッシュの銅グリッド上に15分間置き、続いてろ紙で分散液を注意深く除去することによって準備されました。 SEM測定用のサンプルは、純粋なPSまたはナノコンポジットの透析水分散液5μlをガラス板に滴下することによって準備されました。乾燥したサンプルは、薄い(〜7 nm)金の層でスパッタコーティングされました。
合成された材料の熱安定性は、10°C /分の加熱速度でMOMQ-1500 D(Paulik-Paulik-Erdey)デリバトグラフシステムを使用した場合の空気中のサンプルの熱重量分析(TGA)によって研究されました。
>合成されたナノコンポジットの導電特性を特徴づけるために、それらの粉末は、240°C、5 MPaで2分間の圧縮成形技術(SPECACプレスを使用)と、超音波処理。
合成されたPS / PANI-LSAナノコンポジットの有害ガスに敏感な材料としての適用性を推定するために、最も導電性の高いナノコンポジットNC15を使用し、同じ条件下で合成された純粋なPANI-LSAとその特性を比較しました。アンモニア濃度が19〜152 ppmの範囲のアンモニアと空気の混合物は、分析物として機能しました。敏感な要素は次のように準備されました。溶媒中のナノコンポジットの超音波処理された分散液の1μL容量(2% w / v )は、ガラスセラミック基板上に形成された金の交互嵌合電極のミニチュアシステムにドロップキャストされました。形成された検知素子は、60°Cで30分間乾燥された後、他の場所で説明されている気密試験室に設置されました[30]。調製されたアンモニアと空気の混合物は、このチャンバーに注射器で注入されました。これらの要素のセンサー応答(SR)は、周囲温度と相対湿度約50%で記録され、抵抗の相対変動として決定されました R 方程式SR =[( R に従って、分析物にさらされたセンサーの − R 0 )/ R 0 ]×100%、ここで R はサンプル抵抗、 R 0 は初期抵抗値です。
結果と考察
合成されたPS / PANI-LSAナノコンポジットの形態
TEM画像(図1a)からわかるように、使用された合成アプローチにより、15〜30nmの範囲の非常に小さいサイズの球状のPSナノ粒子を合成することができました。私たちの知る限り、これらのPSナノ粒子は最も低いPSナノ粒子の1つです。

TEM( a – f )およびSEM( g – o )純粋なPSおよびPS / PANI-LSAナノコンポジットの画像: a 、 g -純粋なPS; b 、 h -NC2; c 、 i -NC3; d 、 m -NC6; e 、 n -NC11および f 、 o -NC15
重合後に分離されたPS / PANI-LSAナノ粒子のTEM画像は、PANI-LSA含有量とともにサイズが増加することを示しています(図1b–f)。この効果は、PSナノ粒子のコアとPANI-LSAのシェルを備えたこれらのナノ粒子のコアシェル形態を示唆しています。それにもかかわらず、サイズが大きくなったにもかかわらず、ナノコンポジット(NC2、NC3、NC6)のPANI-LSAの含有量が少ない場合、薄いPANI-LSAシェルを視覚的に区別することは非常に困難です(図1b–d)。この問題は、おそらく両方のコンポーネントのポリマーの性質とこれらのシェルの緩い構造によって説明できます。後者は、次に、コンパクトなPANI-LSAシェルの形成を妨げる大きなサイズのドーパントアニオンによって引き起こされる可能性があります。ただし、NC11、特にNC15のPANI-LSA含有量が高い場合、不規則なシェルを区別できます(図1e、f)。
ナノコンポジット粒子のサイズ分布は非常に広いにもかかわらず(図1b–f)、シェルの厚さを概算できます。特に、PANI-LSAの含有量が最も少ないNC2(表1)には、親PSと同様のサイズが約15 nmのナノ粒子が含まれていますが、TEM画像(図1b)では最大40nmのナノ粒子を見つけることができます。これはおそらく、表面に最大10nmの厚さのPANI-LSAシェルが存在することを示しています。
NC3の場合、15 nmのナノ粒子は観察されませんが、シェルの厚さが最大10 nmの30〜40 nmのナノ粒子の数が大幅に増加しました(図1c)。この傾向はNC6ナノ粒子で強化されています(図1d)。 NC11、特にNC15のTEM画像は、約25〜50 nmの範囲でサイズが大きくなったナノ粒子を示しています(図1e、f)。不規則な形状のいくつかのスポットの存在は、その高い含有量のために、これらのナノコンポジットにPANI-LSAの別の相が現れることを示唆しています。さらに、NC15画像を使用すると、厚さが10〜20nmの不規則なPANIシェルを明確に区別できます。
SEMイメージング用の親PSラテックスの洗浄と準備(「特性評価」のセクションを参照)の後、PSナノ粒子は、おそらく2〜5以上の初期ナノ粒子を含む30〜150 nm以上の範囲のサイズの凝集体を形成しました(図。 1g)。界面活性LSAの存在下でのラテックス培地でのアニリン重合は状況を変えました(図1h–o)。したがって、PANI-LSAの含有量が最も低い場合(1.84 wt%)、NC2画像で、表面が非常に滑らかな約400〜500 nmのサイズの大きな不規則なエンティティを確認できます(図1h)。 PANI-LSA含有量が増加したNC3の場合(3.01 wt%)、エンティティは約100〜300nmの範囲でサイズが小さくなる傾向があります。この傾向は、NC6のPANI-LSA含有量が高いほど強くなります(5.85 wt%)。特に、そのSEM画像は、150 nmまでのサイズの少数のエンティティだけでなく、40〜100 nmの範囲のサイズの不規則な凝集体も示しています(図1m)。 NC11およびNC15のSEM画像(図1n、o)は、サンプルの形態のさらなる発達、つまり、PANI-LSAの含有量がそれぞれ11.27および14.82 wt%と最も高いため、これらのナノコンポジットの質的および量的変化を示しています。具体的には、平らなNC11サンプル表面に主に約25〜50 nmの範囲のサイズの非常に密集した凝集体を見ることができますが、NC15サンプルの場合、「ブドウの房」に配置された25〜50nmの凝集体をよく区別できます。形態が観察されます(図1n、o)。この形態は、他のナノコンポジットと比較して、NC15の比表面積が高いことを示唆しています。
一般に、TEMおよびSEM測定は、洗浄後の純粋なPSナノ粒子は凝集する傾向がありますが、ナノコンポジットNC2およびNC3のPANI-LSAの含有量が少ないとこの凝集が抑制されることを示しています。この効果は、PSコア上の正に帯電したPANIシェルの周囲に局在し、したがってナノ粒子を分離する、電荷を補償する大きなLS¯アニオンの表面活性に割り当てることができます。ただし、中程度(NC6)、特に高(NC11およびNC15)のPANI-LSA含有量では状況が逆転し、PSコアの周りに非常に厚いPANI-LSAシェルの形成が明らかに促進されます。その結果、電荷補償LS ¯ の数 正に帯電したPANIシェルの周囲と内部の両方の陰イオンは、NC2およびNC3の場合と比較して高くなります。必然的に、長いドデシルテールを持つこれらの両親媒性アニオンは、システムの分子間相互作用に存在することを強化することができます。これらの相互作用は、NC2およびNC3での上記の傾向よりもおそらく強力であり、NC6、NC11、およびNC15ナノ粒子の観察された凝集を引き起こす可能性があります。
FTIR測定
合成されたポリマーの構造は、FTIRスペクトルによって特徴付けられます。特に、図2に示すように、PSのFTIRスペクトルには、芳香族C–H伸縮振動の5つの特徴的なピークが含まれ、最大ピークは3025 cm -1 です。 [31]。メチレン基のC–H伸縮振動のピークは、2920および2850 cm -1 で発生します。 。 1601、1583、1492、および1452 cm -1 で、芳香族C =C伸縮振動の4つのバンドが観察されます。 。 756および697cm -1 の非常に強いバンド CHの面外振動とリングの面外変形にそれぞれ割り当てることができます[31]。これらのバンドは、一置換芳香族基の存在を確認します。
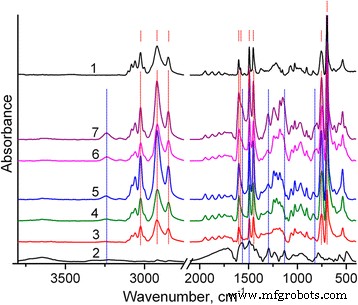
PS(1)、PANI(2)、およびPS / PANI-LSA複合材料のFTIRスペクトル:NC3(3)、NC3(4)、NC3.5(5)、NC11(6)、NC15(7)。 PSとPANI-LSAの主な特徴的なピークは、赤の破線でマークされています。 および青い線 、 それぞれ。すべてのマークは、テキストで説明されている頻度に対応しています
同様に、PANI-LSAのFTIRスペクトルは、公開されているデータとよく一致しています[32、33、34]。 1565、1492、1294、1133、および818 cm -1 の典型的なバンドが含まれています キノイド環、ベンゼノイド環の伸縮振動、二級芳香族アミンのC–N伸縮、B–NH + の振動モードに割り当てられます = Q 構造、それぞれ1.4リングのC–H面外曲げ。 3l00〜3500 cm -1 の領域での非常に弱いNH伸縮振動など、一部の機能 、PANIがドープ状態にあることを示します。ただし、B–NH + = Q 1133 cm -1 でのバンド強度 このPANI-LSAのドーピングレベルが非常に低いことを示唆する非常に弱い[34]。
約1180cm -1 の明確なバンド (図2および3b、曲線1)S =O伸縮振動[31]に由来することは、合成されたPSナノ粒子がラウリル硫酸アニオンを含むことを示しています。さらに、これらの陰イオンの過剰は、最終的なPS / PANI-LSA複合材料で明らかに観察されます。したがって、図3a(曲線2)からわかるように、PANI-LSAの芳香環とメチレン基のC–H伸縮振動は非常に弱いです。したがって、メチレン基のC–H伸縮振動の強いバンドは、NC15スペクトルからPSスペクトルを差し引いたために明らかになります(バンドの高さ3025 cm -1 で正規化した後)。 )(図3a、曲線4)は、明らかに別のSLSフェーズに割り当てることができます。
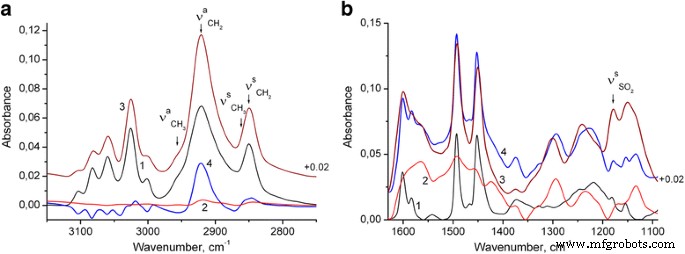
PSナノ粒子(1)、PANI-LSA(2)、およびNC15(3)のFTIRスペクトル: a スペクトル4は、NC15スペクトル b から正規化されたPSスペクトルを差し引いた結果です。 スペクトル4は、PSスペクトルの合計です(3025 cm -1 でのNC15のバンド高さに正規化されています。 )、およびPANI-LSAスペクトル(1560 cm -1 でのNC15のバンド高さに正規化) )
ナノコンポジットのPANIの状態を評価するために、NC15のスペクトルをモデルスペクトルと比較しました(図3b、それに応じて曲線3および4)。最後は、PSとPANI-LSAのスペクトル寄与の合計です。特に、PSの寄与は、3025 cm -1 でのNC15のバンド高さに正規化されたPSスペクトルです。 (PANI-LSA吸収が非常に弱い場合)、PANIの寄与は、1560 cm -1 でのNC15のバンド高さに正規化されたPANIスペクトルです。 (PS吸収がない場合)。約1580および1490cm -1 にドープされたPANIバンドが知られています。 それぞれキノイド環とベンゼノイド環からの主要な寄与があります[32、33、34]。これらのバンドの強度比はPANIの化学構造に敏感であるため、モデルスペクトルと比較したNC15のスペクトルのベンゼノイドユニットに対するキノイドリングの優位性は、PANI-LSA相の酸化度がナノコンポジットは純粋なPANIよりも高いです。また、1601および1583 cm -1 での芳香族C =C伸縮振動のPSバンドもわかります。 NC15スペクトルで広がり、わずかに低波長にシフトします。このシフトは、おそらくPANIとPSの間のπ–π相互作用を示しています。 1133 cm -1 でのNC15バンドの強度 はモデルスペクトルよりもかなり高く、純粋なPANIと比較してこのナノコンポジットのPANI相の導電率が高いことを示しています。
熱安定性
最近、PANIの含有量が非常に少ない(PANIベースの約2 wt%、または異なる芳香族スルホン酸をドープした場合は3.5〜5.0 wt%)ポリカーボネート(PC)のミクロンサイズの粒子状コアシェルポリマー-ポリマー複合材料で示されています-ドーパント)ドーパントの存在がそれらの熱安定性に強く影響する[35]。ドーパントの芳香環のアルキル置換基に応じて、複合材料は、可塑化する大きなドーパントアニオンの特定の分子間相互作用および/またはPC鎖を持つ熱放出ドーパント分子[35]。ただし、シェル内のPANIが通常デドープ(ベース)状態にある500°Cを超える温度では、複合材料の安定性はPCの安定性よりも高くなりました。この効果は、コアシェル複合材料のコア材料の表面にシェルとして配置されたPANIの特定の状態と、PANIシェルによるPCコア粒子の安定化の可能性に割り当てられました[35、36]。この可能性とナノコンポジットの形態に基づいて、PANIシェルの安定化効果はさまざまなポリマー-PANIコア-シェルナノコンポジットでも発生する可能性があることを示唆しています。この提案は、図4に示す合成PSナノ粒子およびナノコンポジットの熱的挙動とよく一致しています。
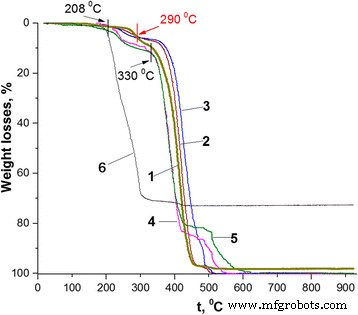
異なるPANI-LSA含有量(wt%)のPS / PANI-LSAナノコンポジットの熱重量曲線: 1 PS、 2 1.84(NC2)、 3 3.01(NC3)、 4 11.27(NC11)、 5 14.82(NC15)、 6 SLS
実際、合成されたPSナノ粒子は、バルクPSとはある程度異なる熱安定性を示します(図4の曲線1と[37]の図1を比較してください)。特に、後者は主に200〜450°Cの単一ステップで空気中で分解しますが[37]、前者の熱重量(TG)曲線は、最初から1週間の体重減少(〜1.9 wt%)のおよそ3つの段階を示しています。 262°C、2番目は262〜330°Cの範囲、3番目は330〜505°Cの範囲です。この違いは、PSナノ粒子合成の特異性により、SLS不純物の組成に不可避の存在が生じ、PSの熱的挙動が変化したことで説明できると考えられます。この提案は、SLSの最終分解温度がPSナノ粒子分解の第3(メイン)段階の開始(330°C)に非常に近いという事実とよく一致しています(図4、曲線1および6)。
図4に示すように、ナノコンポジットのTG曲線は、PSの形状と類似しており、さらに、120°Cまでの温度で同様の小さな質量損失を示します。これは通常、水分蒸発に割り当てることができます[35]。 。高温では、PANI-LSAの負荷が低い場合と高い場合で、サンプルの熱安定性に大きな違いが見られます。これにより、一般に、既知のコアシェルPANI含有複合材料の熱挙動の特異性に関する補足情報が得られます。特に、PANI-LSA含有量が11.27 wt%以下の3つのナノコンポジット(NC2、NC3、およびNC11)は、208°CまでのPSの高い熱安定性と同様に表示されます(図4、表2)。ただし、PANI-LSA含有量が最も高い(14.82 wt%)NC15は、120°CでもPSよりも安定性が低く(図4、曲線1および5、表2)、おそらく水分だけでなく蒸発にも割り当てることができます。しかし、おそらく未結合のドーパントおよび/または未反応のモノマー/オリゴマー不純物にも影響します[38]。
<図>208〜262°Cの温度範囲では、すべてのナノコンポジットが重量損失を示します。これは、LSA含有量よりも高いですが、PSの重量損失よりも大幅に少ないです(図4、曲線1、4、および5、表2)。ただし、NC2およびNC3の場合、これらの損失はPANI-LSAの内容よりもさらに高くなります。 PANIベースの高い熱安定性[39]とPSナノ粒子の熱的挙動(図4、曲線1)に基づいて、ナノコンポジットの損失をドーパントの蒸発と分解だけでなく、 PSコンポーネント。さらに、262°CでのNC2とNC3の重量損失(表2)は、LSA含有量とPS損失の合計(それぞれ3.02と3.7)を上回っていますが、PSコアコンポーネントの熱酸化劣化がある程度強化されていると考えられます。ナノコンポジットは、ドーパントの分解生成物によって引き起こされる可能性があります。
ナノコンポジットの損失は通常、高温で増加しますが、290°Cでは、NC2とNC3のTG曲線(NC11とNC15のTG曲線とは異なります)は、5.58 wt%の点でPSのTG曲線と交差します(図4、表2)。 。この振る舞いは、一般に、ドーパントの完全な喪失[35、37、38]と、脱ドープされたPANIのPANI-LSA成分の変換を示唆しています。これを超えると、加熱プロセスが終了するまで、温度NC2およびNC3はPSよりも安定します(図4、曲線1〜3)。 As a consequence, the position of the TG trace of PS nanoparticles along the temperature axis in the range of 262–430 °C roughly separates positions of the nanocomposites with low and high contents of PANI-LSA (Fig. 4). This fact confirms a difference which is probably inherent to these two sets of nanocomposites.
Indeed, one can see strongly different course of the thermal degradation of these nanocomposites both in the range of 262–430 °C and above 430 °C. Whereas all these nanocomposites have the core-shell morphology, it is unlikely that only this morphological factor can explain their specific thermal behavior. However, if to take into account the presence in their composition of the LSA dopant, which contains the long dodecyl tail with plasticizing ability [3], we can at least partially understand such difference as a result of intermolecular interactions (causing a plasticizing effect [40]) of the dopant anion with the polymer components of the nanocomposites. Naturally, in the case of low or high contents of the PANI-LSA component, its influence on thermal behavior of the nanocomposites will be less (NC2, NC3) or more (NC11, NC15) significant. In the latter case, the plasticization effect is so strong that the thermogramms of NC11 and NC15 (Fig. 4, TG curves 4 and 5) take positions below the PS one up to 430 °C even after a complete removal of the dopant (above ~ 290 °C) because of weakened interactions between PS macromolecules. Slowing down the degradation rate of the nanocomposites with high content of PANI base at temperatures above 430 °C can be probably explained by cross-linking of its chains [39] and possible enhancement of the stabilizing role of the PANI shell.
In the case of NC2 and NC3, the situation is obviously opposite to NC11 and NC15. In particular, contents of PANI-LSA are quite small, and therefore, quantities of the plasticizing dopant LSA are not enough to significantly weaken interactions between PS macromolecules. As a consequence, once the dopant is eliminated completely, the nanocomposites display thermostability which is higher than that of PS nanoparticles (Fig. 4, curves 2 and 3, intersection point at 290 °C). In spite of the low content of PANI-LSA and, therefore, of its thin shell, these NC2 and NC3 behaviors match well with the suggestion about stabilizing effect of the PANI base shell.
Conductivity and sensing properties of the synthesized nanocomposites
One of most important features of polymer-polymer composites, in particular of PANI-containing composites, is probably their ability to withstand conditions of common treatments, which are typically applied to produce different articles. Therefore, a lot of studies have been performed to estimate changes in properties of these materials after treatments by melting or solution techniques [3, 38, 39]. Based on these studies and the thermally induced weight losses of the synthesized nanocomposites (Table 2, Fig. 4), one might expect that such important property of doped PANI as conductivity could be changed under these treatments. Indeed, as one can see from Fig. 5, the values of conductivity of the cast and compression molded PS/PANI-LSA films strongly differ. To quantify the difference, we treated these data (Fig. 5) by the scaling law based on the percolation theory [41] in accordance with the known methodology of processing the conductivity behavior of polyaniline networks in composites [42, 43]:
$$ \sigma ={\sigma}_{\mathrm{o}}{\left(f-{f}_c\right)}^t $$ (1)ここで、σ o is the constant displaying conductivity of the PANI conducting phase, f is the volume fraction of PANI, f c is the percolation threshold, and t is the critical exponent. Volume fractions of PANI-LSA in the nanocomposites were calculated on the basis of densities of PS and PANI-LSA, i.e., 1.04 [44] and 1.18 g/cm 3 [45], respectively.

Dependencies of DC conductivity of the cast (1 ) and compression molded (2 ) PS/PANI-LSA nanocomposite films on the volume fraction of PANI-LSA
The power-law dependence was determined with various trial values of f c by applying a linear regression analysis to the plot of log σ versus log (f − f c )。 The solid lines represent best fits to the data with the correlation coefficients of 0.996 and 0.993 for the cast and compression molded nanocomposite films, respectively.
The observed nonlinear dependences (Fig. 5) are obviously the result of formation of the phase-separated conducting percolation network of PANI-LSA in the bulk of the nanocomposite films. It is interesting to note that the percolation thresholds are quite low (f c = 1.26%) and independent on the used processing techniques. This f c value is significantly lower than the theoretical model suggests for a random lattice of spheres (from 15 to 30% depending on the sphere diameter) [41]. However, the conductivity of the PANI conducting phase (σ o ) in the cast nanocomposite films is more than two and a half times higher than that of the compression molded ones (2.3 × 10 −4 and 8.9 × 10 −5 S/cm, respectively). Obviously, the lower conductivity of the conducting phase in the compression molded film is caused by the partial thermal degradation of PANI-LSA under the melting treatment temperature (240 °C). The values of the critical exponent t for the cast and compression molded films are 1.14 and 2.62 accordingly. Such inequality in the critical exponent indicates a strong difference in the spatial structure of the percolation cluster, which results in the different slopes of the curves. As a consequence, the conductivities of the cast nanocomposite films are more than three orders of magnitude higher than those of the compression molded ones at low volume fractions (contents) of PANI-LSA (Fig. 5).
Nevertheless, despite the significant difference of the cast and compression molded films, one can deduce that the conductivity level is enough to apply the both materials for antistatic applications. On the other hand, the obtained conductivity values of the synthesized nanocomposites are significantly lower (by 2–3 orders of magnitude) than in the case of the similar core-shell submicron/micron-sized PS/PANI composites [4, 7, 9, 14]. To understand this difference and to improve the conductivity of these new PS/PANI-LSA nanocomposites, new studies are planned. However, one can suggest that non-optimal conditions of preparation of these new materials are at least partial explanation of this low conductivity level.
Based on better conductivity properties of the cast PS/PANI-LSA films and known high sensing ability of doped PANI [46], we estimated their potential as sensing materials to determine concentrations of ammonia in its gaseous mixtures with air. The measurements were performed on the example of the films of NC15 and pure PANI-LSA cast on electrodes of the transducer (see “Methods” chapter).
Both films demonstrate quite high sensitivity to ammonia in the range of 19–152 ppm (Fig. 6). However, while NC15 is more sensitive to ammonia than pure PANI-LSA in the concentration range of 19–114 ppm, at higher concentrations, the situation becomes opposite.

Sensor responses (calibration curves) of the cast pure PANI-LSA (1 ) and NC15 (2 ) films to different concentrations of ammonia in the mixtures with air
The better efficiency of NC15 in this narrowed ammonia concentration range can be probably assigned to core-shell morphology of the nanoparticles constituting the cast nanocomposite film. This morphology typically specifies higher surface of the film as compared with pure PANI-LSA and improves sensitivity of sensing materials [25, 28]. The enhancement of the sensing responses of pure PANI-LSA at ammonia concentrations above 114 ppm (Fig. 6, curve 2) can be probably assigned to additional involving in the sensing process of the PANI-LSA clusters located under the surface of the film. Naturally, the quantity of these clusters in the pure doped PANI film is much higher than in the case of the thin PANI-LSA shells on the core particles constituting the nanocomposite film. Therefore, their involvement in the sensing process inevitably increases sensor responses of the pure doped PANI film as compared with the NC15 one.
Conclusions
The new PS/PANI-LSA nanocomposites have been synthesized with the core-shell nanoparticle sizes ~ 25–50 nm, which to our knowledge are the lowest ones among the similar composites published elsewhere. The use of LSA as acidifying agent for the aniline containing PS latex medium and addition of the oxidant resulted in the precipitation of the thin PANI-LSA shell (~ 10–20 nm) on the surface of the PS nanoparticles (synthesized in the presence of SLS). As a consequence, both the shell and PS core contained the same lauryl sulfate surface active anion unlike the known core-shell PS/PANI composites synthesized with a PS latex surfactant-stabilizer and PANI dopant of different nature.
We have found that although the synthesized very small PS nanoparticles (15–30 nm) after cleaning tend to agglomerate in the dry state, the low contents of PANI-LSA in the nanocomposites suppress this agglomeration probably due to the surface activity of charge compensating large LS¯ anions which localize around positively charged PANI shells on PS cores and separate the nanoparticles. However, the situation becomes opposite at the moderate and especially at the high PANI-LSA contents, which apparently facilitate formation of quite thick PANI-LSA shells around PS cores. In this case, a number of charge compensating LS ¯ anions both around and inside of the positively charged PANI shells is higher as compared with the low contents of PANI-LSA in the nanocomposites. As a consequence, these amphiphylic anions with long dodecyl tails can enhance intermolecular interactions in the system and lead to the agglomeration of the nanoparticles with high contents of PANI-LSA.
A possibility of such agglomeration effects should be taken into account when using similar nanocomposites in applications which need charged nanoparticles [7, 20, 25]. We believe that applied in this work method of determination of the real PANI content in the PS/PANI nanocomposites can allow better control of their properties.
Based on FTIR and conductivity studies of the synthesized nanocomposites, we proved that oxidation state and conductivity of the PANI phase are appreciably higher than those of pure PANI-LSA. Moreover, we demonstrate here that thermal behavior of these nanocomposites in air is strongly different for low and high PANI-LSA loadings that probably stems both from the plasticizing ability of the LSA dopant and stabilizing effect of the PANI shell. This fact, in general, gives the complementary information about the thermal behavior specificity of the known PANI containing core-shell composites.
At the same time, based on thermal stability, conductivity and sensor studies, we conclude that properties of the synthesized PS/PANI-LSA nanocomposites testify to their potential applicability as materials for antistatic and sensing applications.
ナノマテリアル
- ACEO®がシリコーンを使用した3Dプリントの新技術を発表
- 3Dプリントで航空宇宙を新たな高みへ(2020)
- Computer-on-Modulesは新しいOSM標準でミニチュアになります
- congatec:NXP i.MX8Mミニプロセッサを搭載した新しいSMARCモジュール
- 新しいドラフト樹脂を使用した迅速なSLAプロトタイピング
- 何か新しいもの:IMLを使用したPLAのコーヒーカプセル
- Honeywellは、新しいパートナーシップでエネルギーIIoTを強化します
- オムロンがAIを内蔵した新しい産業用ロボットを展示
- IoTを使用した5G:デジタル化の新時代
- 新しいEDMテクノロジーでニッチを探る
- 新しいリソースを使用すると、アセットタグの選択が簡単になります