


クリーンでグラフェンで覆われたCu(111)表面へのバナジウム原子の吸着の電子的性質
要約
きれいでグラフェンで覆われたCu(111)表面に吸着されたバナジウム原子の電子特性は、abinitio理論的方法を使用して体系的に研究されています。この作業では、バナジウム吸着の2つのカバレッジ(1 / 9MLと1ML)が考慮されます。私たちの計算によると、Cu表面の下にとどまるVは、前述のV / Cu(111)の2つの被覆率で最も安定した吸着サイトであることがわかります。しかしながら、そのような吸着は望ましくない特性につながる可能性があります。したがって、VとCu表面間の直接相互作用を効果的に軽減するために、バッファ層としてグラフェンを導入します。計算は、元のグラフェン層の電子特性が、C原子とV吸着原子の相互作用によって大きく影響を受けることを示しています。グラフェンのディラックポイントは、両方のカバレッジで結果として「破壊」されます。 V / Gra / Cu(111)システムでは、Gra / Cu(111)システムと同様に、グラフェン層と基板のCu原子間の相互作用は弱いままです。さらに、1/9 MLの比較的低いカバレッジは、スピン偏極システムを生じさせますが、非スピン偏極システムは、1MLのカバレッジで観察されます。この発見は、バナジウムベースの材料を実際に適用するための新しい方法を提供します。
背景
不均一系触媒作用は、化学およびエネルギー産業の多くの分野で重要な役割を果たします。これまで、集中的な研究は、新しい触媒の理解、改善、および設計に焦点を合わせてきました。貴金属基板への遷移金属原子の吸着は、対応する触媒特性に影響を与える可能性があります。これは、触媒作用における最も重要なトピックの1つです[1,2,3,4,5,6,7]。特に、金属表面への1つの単分子層金属の吸着は、さまざまな種類の吸着システム内で著しく異なる化学的および触媒的特性を示します[5、6、7]。一般に、材料の触媒特性は、それらの原子構造、組成、およびフェルミ準位に近い電子状態に依存します[8、9、10、11、12]。基板は、金属堆積物の触媒特性に直接的および/または間接的に影響を与えると予想されます。ご存知のように、Cu(111)表面は、過去数十年間で最も徹底的に調査された単結晶金属表面の1つです[13、14、15、16、17、18、19、20、21、22、23、24 ]。特に、過去10年間で、Cu(111)表面は、化学蒸着(CVD)による高品質で大面積のグラフェンの成長のための最も主要な基板と見なされてきました[22、23、24]。グラフェンの新しい電子特性は、このような基板上に十分に保存できます。 Cu(111)表面への後期4d遷移金属(Rh [25]、Pd [26,27,28,29,30]、Ir [31]、Pt [29、32、33]など)の吸着実験的にも理論的にも広く研究されてきました。しかし、Cu(111)表面に吸着された初期の3d遷移金属原子の研究は比較的不足しています[34,35,36,37]。ここでは、初期の3d遷移金属元素であるバナジウムに焦点を当てます。これは、その生化学的関連性と、不均一系触媒作用、分子ネットワーク、ナノ材料、電池の構築など、いくつかの産業分野での広範なアプリケーションによるものです[38]。バナジウムベースのポリアニオン材料は、市販のカソード材料LiCoO 2 に代わる候補として提案されています。 およびLiMn 2 O 4 その柔軟な原子価状態のために[39]。したがって、バナジウム原子の吸着特性を研究することで、実際の応用が容易になります。調査したシステムの可能なアプリケーションは、以下のように期待できます。 (1)バナジウムの一般的な酸化状態は、+ 2、+ 3、+ 4、および+5です。したがって、ナノマテリアル産業で強力で用途の広い触媒として使用できます[38]。 (2)金属状態のバナジウムは、COからCおよびCO 2 への不均化を触媒するために使用できます。 [40]。 (3)電気伝導と熱伝導が増加する可能性があるために自由電子の濃度が弱い表面に吸着されたTM(つまりバナジウム)原子を分析することも興味深いです[41]。さらに、記録媒体、磁気インク、およびスピントロニクスデバイスで使用できる2次元表面システムの磁気秩序に非常に関心があります。
この作業では、密度汎関数理論(DFT)に基づいて、きれいなCu(111)表面とグラフェンで覆われたCu(111)表面へのバナジウム原子の吸着の体系的な調査を報告します。上記の2つのシステムでは、バナジウム吸着原子の2つの対照的なカバレッジ(つまり、1 / 9MLと1ML)を考慮して、電子的および磁気的特性に対するカバレッジの影響を評価します。きれいなCu(111)表面に吸着されたVの最低エネルギー吸着サイトは、Vの被覆率に関係なく、表面の上ではなく表面の下にあります。グラフェンで覆われたCu(111)表面へのVの吸着では、吸着サイトはカバレッジに依存します。つまり、最大の配位を持つ中空サイトが1/9 MLのカバレッジに対してエネルギー的に優先され、一方、低い配位のトップサイトがエネルギー的に優先されます。 1MLのカバレッジに適しています。一方、V / Cu(111)システムとV / Gra / Cu(111)システムの両方で、V吸着原子のスピン分極は1/9 MLカバレッジでエネルギー的に有利ですが、1MLカバレッジでは磁性は見つかりません。さらに、グラフェンのC原子の正味の磁気モーメントは約0.16μ B です。 / 9 ML V / Gra / Cu(111)システムの炭素あたり。これは、Gra / Cu(111)システムの結果とは異なります。 V / Cu(111)およびV /グラフェン/ Cu(111)システムの相互作用を深く理解するために、フェルミ面の電子状態を詳細に分析します。簡単に言えば、私たちの研究は、V / Cu(111)およびV / Gra / Cu(111)システムの電子特性を理解するのに役立つ可能性があります。
メソッド
私たちの計算は、スピン偏極密度汎関数理論[43]、平面波基底、およびプロジェクター拡張波(PAW)表現[44]に基づくVienna ab initioシミュレーションパッケージ(VASP)[42]を使用して実行されました。 ]。一般化勾配近似(GGA)内のPerdew-Burke-Ernzerhof(PBE)交換相関エネルギー汎関数[45]が計算に使用されます(B3LYP [46、47]およびHSE06 [48]混成汎関数を使用したいくつかの比較研究も必要に応じて提示)。グラフェンとCu(111)表面の間のファンデルワールス(vdWs)相互作用を正確に記述するために、vdWs補正を使用したPBE汎関数(DFT-D2)[49]が採用されています。カットオフ平面波の運動エネルギーは500eVに設定されています。 Cu(111)表面は、約20Åの真空間隔で7つのCu層を含むスラブモデルを使用してモデル化されました。 Cu(111)およびGra / Cu(111)表面の異なるVカバレッジは、異なるスーパーセルを使用してモデル化されました。 1 / 9MLと1MLのVカバレッジでは、それぞれ(3×3)と(1×1)の表面ユニットセルを使用しました。 24×24×1k -のMonkhorst-Packスキーム[50] メッシュを使用して、(1×1)表面ユニットセルのブリルアンゾーン積分をサンプリングし、8×8×1k - (3×3)表面ユニットセルにはメッシュを使用しました。最適化中、スラブの最下部の3つのCu層は凍結され、システムの残りの原子は、各原子にかかる力が0.01eV /Å未満になるまで完全に緩和されました。バナジウム原子はスラブの片側に吸着されました。双極子補正[51]は、計算に基づいて検出されたエネルギー補正が無視できるため、この研究では考慮されていません。
結果と考察
きれいなCu(111)表面へのバナジウム原子の吸着
このセクションでは、2つのカバレッジ(つまり、1 / 9MLと1ML)でのクリーンなCu(111)表面へのV吸着の結果を示します。 Cu(111)表面上のV原子の好ましい吸着サイトを見つけるために、各カバレッジに対して7つの可能な吸着サイト、つまり、top、fcc、hcp、subT、top-fcc、top-hcp、およびbridgeサイトが考慮されます。 、図1aに示すように。特に、subTはCu表面の下のサイトであり、V吸着原子はその位置を表面層のCu原子と交換します(そしてCu原子をバナジウム原子の真上のサイトに移動します)。図2a、bを参照してください。両方の吸着範囲について、7つの吸着サイトすべての吸着エネルギーがV / Cu(111)システムに対して計算されます。得られた結果を図1b、cに示します。ここで、バナジウム原子あたりの吸着エネルギー(E ad )は次の式で計算されます:
$$ {\ mathrm {E}} _ {\ mathrm {ad}} =\ left [\ left({NE} _V + {E} _ {Cu(111)} \ right)-{E} _ {V / Cu (111)} \ right] / N $$ここで E V は、孤立したバナジウム原子 E のエネルギーです。 Cu (111) 関係するクリーンなCu(111)表面の総エネルギー E V / Cu (111) はV / Cu(111)システムの総エネルギーであり、 N 関与するV原子の数です。図1b、cから、前述の被覆率のCu(111)表面へのV吸着にはsubTサイトがエネルギー的に有利であることがわかります。これに基づいて、以下の説明ではsubTサイトのみを検討します。計算された吸着エネルギー、V原子とその隣接するCu原子間の結合長、およびV / Cu(111)のV原子の原子磁気モーメントを表1に示します。表1からわかるように、吸着エネルギーはE ad 1 / 9MLおよび1MLカバレッジの場合、V原子あたりそれぞれ2.17および1.61 eVです。これは、V原子とCu(111)表面との相互作用が非常に強いことを示しています。さらに、吸着エネルギーはV被覆率の増加とともに減少します。これは、V-V相互作用が強くなり、V層とCu表面間の相互作用が弱くなることを意味します。 V原子とその隣接するCu原子の間の最短結合長は、1 / 9MLと1MLのカバレッジでそれぞれ2.27Åと2.37Åです。これは、V吸着原子とCu基板の間の相互作用が、1/9 MLで比較的強いことを意味します。これは、吸着エネルギーから計算された結果と一致しています。 V吸着原子の強磁性(FM)次数も計算で考慮され、FM次数のスピン偏極エネルギーはΔ E によって計算されます。 =( E いいえ _ mag − E FM )/ N ( E いいえ _ mag 非磁性状態のエネルギーです)。バナジウム原子のスピン偏極エネルギーは、1 / 9MLカバレッジでは110meVですが(表1を参照)、1MLカバレッジでは磁性がありません。 Vの原子磁気モーメントは1.34μ B バナジウムの1 / 9MLカバレッジの場合、値(3μ B )とは大きく異なります。 )気相V原子の。この点については後で説明します。
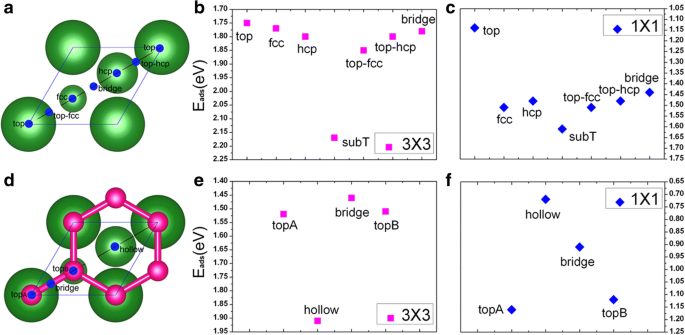
a きれいなCu(111)(1×1)表面の吸着サイト:最大のボールは表面のCu原子を示し、小さいボールはサブレイヤーのCu原子を示します。 b 、 c それぞれ1 / 9MLおよび1MLカバレッジでのCu(111)上のV原子の異なる吸着サイトの吸着エネルギー。 d グラフェンで覆われたCu(111)(1×1)表面、赤いボールはグラフェンのC原子を示します。 e、f それぞれ1 / 9MLおよび1MLカバレッジでのグラフェン被覆Cu(111)上のV原子の吸着エネルギー
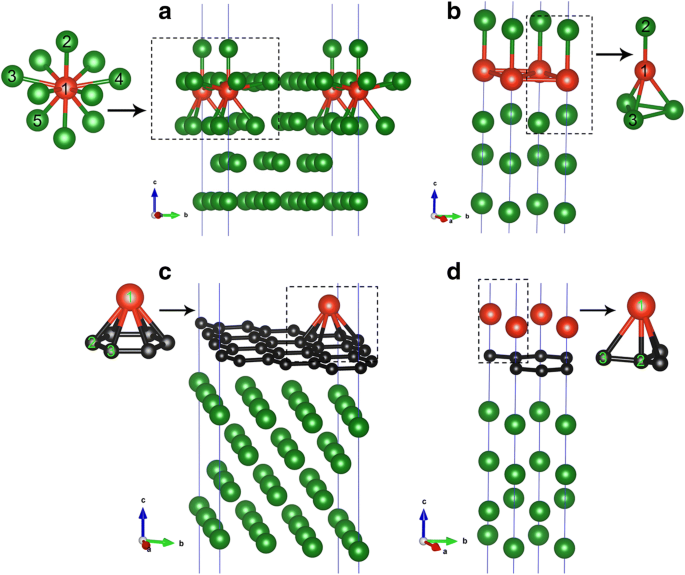
a のV / Cu(111)システムの形状 1 / 9MLおよび b 1MLカバレッジ。 c のV / Gra / Cu(111)システムの形状 1 / 9MLおよび d V原子の1MLカバレッジ。赤、黒、緑のボールはそれぞれV、C、Cu原子を表します
次に、V / Cu(111)システムの電子構造について説明します。 Cu(111)(3×3)およびCu(111)(1×1)表面(すなわち、1/9および1 ML)へのV吸着のバンド構造を図3に示し、対応するクリーンなCu(111)(3×3)およびCu(111)(1×1)表面も比較のためにプロットされています。図3a、dはどちらも、クリーンなCu(111)の電子構造について議論するために使用できます。ここで図3dを選択します。両方の 電子と d Cuの電子は、きれいなCu(111)表面のシステムのコンダクタンスに寄与します。より詳細には、図3dのフェルミ面上の代表点(A、B、C、D、E)にラベルを付けました。ポイントAとEは、主に d から提供されます。 yz 表面のCu原子の電子。ポイントBとCは、 d からの貢献を示しています xy および d x 2 − y 2 それぞれCu原子の電子。ポイントDは、 s の混合について説明しています。 d の電子 z 2 および d x 2 − y 2 隣接するCu原子間の電子。 VがCu(111)に吸着されると、V被覆率が変化するにつれて、得られるバンド構造は異なる方法で変化します。 1/9 MLカバレッジ(図3b、cに表示)の場合、スピンアップチャネルとスピンダウンチャネルのバンド構造が異なり、スピン偏極した特徴を示しています。図3では、赤い点はV吸着原子からの寄与を表し、銀色の灰色の点は背景Cuの寄与を示しています。スピンアップチャネル(つまり、多数回転)から、両方の d V原子と基板のCu原子の電子は、フェルミ面の電子状態に大きく寄与します。 d のハイブリダイゼーション 表面のCu原子とV原子の電子がはっきりと見えます。明確に説明するために、図3bのフェルミ面上のいくつかの代表的な点(A、B、C、D)にもラベルを付けました。その中で、点Aは d の混合を示しています z 2 d のV吸着原子の電子 yz 、 d z 2 表面のCu原子の電子。すべてのB、C、およびDポイントは、 d からの寄与を示します V吸着原子の電子。例として、ポイントBは d のみからの寄与を示しています x 2 -y 2 V吸着原子の電子。スピンダウンチャネル(つまり、少数スピン)の場合、バンド構造は、V吸着原子から寄与した電子状態がすべてフェルミ準位(空いている)をはるかに上回っていることを示しています。フェルミ面への寄与は主に s によるものです 、 d Cu原子の電子、V原子の電子からの寄与はごくわずかです。これら2つのスピンチャネル間に存在する違いは、V吸着原子の磁気モーメント(1.34μ B )を示しています。 )。 1/9 MLの状況とは異なり、1 MLのカバレッジ(図3e、fに示す)の場合、吸着システムはスピン偏極していません。便宜上、図3eのフェルミ面の代表点(A、B、C、D、E、F、G、H)にもラベルを付けました。ポイントAとHはどちらも、 d の寄与に対応しています。 yz Cu原子の最下層の電子。ポイントB、D、E、F、およびGの電子状態は、 d によって提供されます。 V吸着原子の電子。たとえば、 d のみ x 2 − y 2 および d xz のV吸着原子は、それぞれBとDの電子状態に寄与します。フェルミ面のBとDの間の点について、 d の複雑な混合が見つかりました。 s、d を持つV吸着原子の電子 V原子の周りのCu原子の電子。たとえば、点Cの場合、電子構造は s、d の混合によって特徴付けられます。 yz 、 および d z 2 表面層Cuの電子とd z を持つ最上部の副層Cu原子 2 V吸着原子の電子。
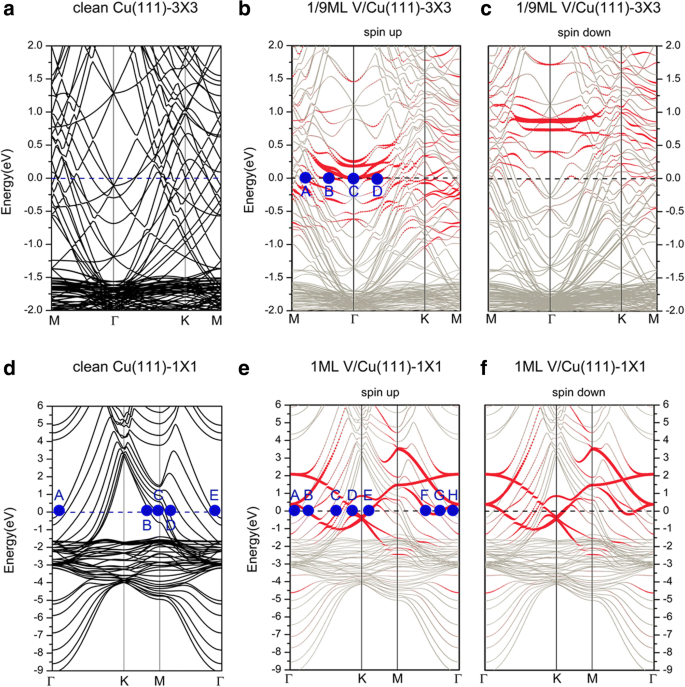
a のブリルアンゾーン(BZ)にプロットされた、きれいなCu(111)表面のバンド構造 3×3ユニットセルおよび d 1×1ユニットセル。 b の1/9 MLカバレッジのCu(111)表面へのV吸着のバンド構造 スピンアップして c スピンダウンします。 e の1MLカバレッジでのCu(111)表面へのV吸着のバンド構造 スピンアップして f スピンダウン
クリーンなCu(111)表面へのV吸着の総状態密度(TDOS)と、投影された状態密度(PDOS)を、1 / 9MLと1MLの両方のカバレッジについて図4に示します。明らかに、V吸着原子の明らかなスピン偏極は1/9 MLで見られますが(図4cを参照)、V吸着原子のスピン偏極は1 MLでは見られません(図4fを参照)。さらに、1 / 9MLと1MLの両方のカバレッジでCu原子のスピン偏極は観察されません(図4b、eを参照)。 V吸着の前後でCuのPDOSを統合することにより(つまり、Cu上の電子の数につながる)、Cu原子の電荷がわずかに増加することがわかりました。これは、V吸着原子からCu基板への電荷移動を示しています。 V / Cu(111)。言い換えれば、V吸着はCuへのn型ドーピングにつながります。 Cu(111)表面へのV吸着をよりよく理解するために、それぞれ1 / 9MLおよび1MLカバレッジでの変形電荷密度の等高線図を図5a、bにプロットします。変形電荷密度は、\(\ Delta \ rho \ left(\ overrightarrow {r} \ right)={\ rho} _ {\ left [V / Cu(111)\ right]} \ left(\ overrightarrow {で定義されます。 r} \ right)-\ sum \ Limits _ {\ mu =1} ^ N {\ rho} ^ {atom} \ left(\ overrightarrow {r}-\ overrightarrow {R _ {\ mu}} \ right)\)。図5に示すように、V吸着原子とその隣接するCu原子の間の共有結合とイオン結合は、1 / 9MLと1MLの両方のカバレッジではっきりと見えます。具体的には、共有結合は1/9 MLカバレッジで比較的強力です(1 MLと比較した場合)が、イオン結合は1MLカバレッジで比較的強力です。
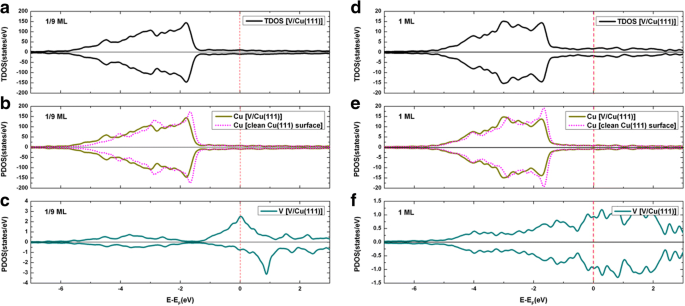
a 1 / 9MLカバレッジのV / Cu(111)システムのTDOS。 b 1 / 9MLでの基板の全体的なCu原子のPDOS。 c 1 / 9MLのV吸着原子用のPDOS。 d 1MLでのV / Cu(111)システムのTDOS; e 1MLでの基板の全体的なCu原子のPDOS。および f 1MLでのV吸着原子のPDOS。特に、きれいなCu(111)表面のDOSもグラフに埋め込まれています b および e 比較のために
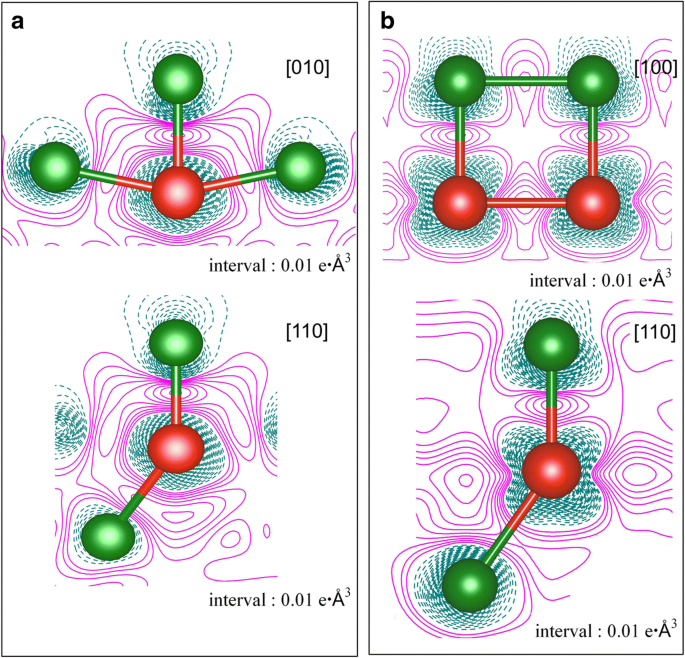
2つのカバレッジ、つまり a でのCu(111)表面へのV原子の吸着の変形電荷密度 1 / 9MLおよび b 1ML。電子の蓄積と枯渇は、それぞれマゼンタの実線と濃いシアンの破線で表されます。緑と赤のボールはそれぞれCu原子とV原子を表しています
Cu(111)表面のグラフェン層
subTサイトは、上記の議論の吸着エネルギーから、クリーンなCu(111)上のV原子の最も安定した吸着サイトであることがわかります。そのような吸着サイトはいくつかの関心事ですが;ただし、Cu表面層の下にとどまるV原子は、表面での触媒としての用途を制限する望ましくない特性につながる可能性があります。したがって、V吸着原子とCu(111)表面の間の直接相互作用を軽減するために、バッファ層の導入を試みます。グラフェンは、この研究で最初に検討されたV / Cu(111)システムとの最も近位の格子整合のために最適な選択です。
金属表面へのグラフェンの吸着は、以前のいくつかの出版物で集中的に研究されてきました[52、53、54]。グラフェンがCu(111)に吸着されると、グラフェン/ Cu(111)システムの3つの可能な形状(以下、Gra / Cu(111)と呼びます)が考慮されました。つまり、グラフェンはtop-fcc、top-hcpにありました。 、およびfcc-hcp吸着サイト。図6a–cを参照してください。私たちの結果に基づいて、top-fccジオメトリ(図6a)は、炭素原子あたり47 meVの吸着エネルギーを持ち、エネルギー的に最も安定した構造であり、グラフェンとCu(111)表面の間の平衡距離は3.14であることが示されています。 Å、これは以前の研究[52,53,54]とかなりよく一致しています。このような低い吸着エネルギー(47 meV / C)と大きな層間距離は、グラフェンとCu(111)間の結合が比較的弱いことを意味します。図6dは、top-fcc構成のグラフェン/ Cu(111)システムのバンド構造を示しています(図6a)。特に、グラフェンから生じる電子状態は、図のマゼンタ色の円でスケッチされています。自立型グラフェンシートのバンド構造も比較のために図6eに示されています。これらの図からわかるように、グラフェンのバンド構造は、独立したシートとCu(111)表面のシートの間で非常に似ています。 Kでのディラック点(線形バンド交差あり)は図6dに保持されていますが、グラフェンがCu(111)表面に吸着されると少しダウンシフトします。交差点のダウンシフトは、Cu(111)基板からグラフェン層への電荷移動を示します。これは、Al、Ag、およびCuがグラフェンによってn型ドープされているという以前の結果と一致しています[52,53,54 ]。変形電荷密度、つまり、Gra / Cu(111)の総電荷密度と、独立したグラフェンとクリーンなCu(111)表面の電荷密度の合計、つまり\(\ Delta \ rho \ left(\ overrightarrow {r} \ right)={\ rho} _ {Gra / Cu(111)} \ left(\ overrightarrow {r} \ right)-{\ rho} _ {Gra} \ left(\ overrightarrow { r} \ right)-{\ rho} _ {Cu(111)} \ left(\ overrightarrow {r} \ right)\)は、図6fにプロットされています。図6fに示すように、C原子の周りの実線の等高線に従って、Cu(111)基板からグラフェン層への電荷移動も観察できます。一方、Gra / Cu(111)系のCおよびCu原子の予測状態密度(PDOS)を、Cu(111)表面の仕事関数変化(グラフェン吸着ありとなし)とともにプロットします。 7. PDOSの積分から、C原子の電子はわずかに増加し、Cu原子の電子はわずかに減少することがわかりました。これにより、電荷移動現象も確認されました。さらに、計算されたCu(111)の仕事関数は、グラフェン層の吸着後に4.78から4.68eVに変化することがわかりました。これらはすべて、電荷移動がCu基板からグラフェン層へのものであることを確認しています。
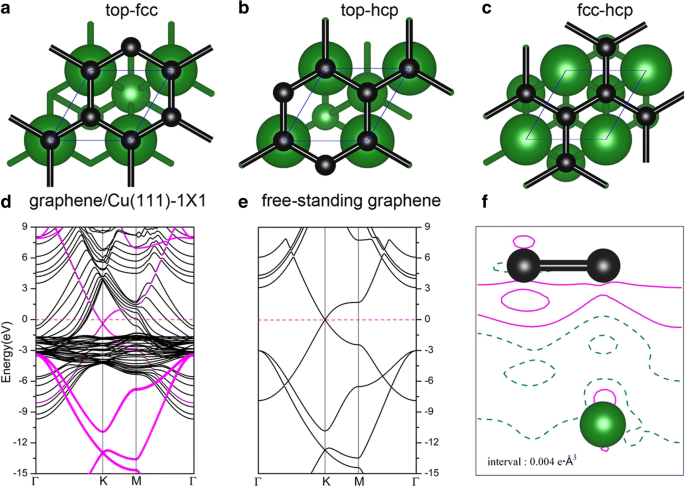
Cu(111)表面のグラフェンシートの形状: a top-fcc、 b top-hcp、および c fcc-hcpサイト。 d e と比較した、top-fccジオメトリのグラフェン/ Cu(111)システムのバンド構造 自立グラフェンのもの。マゼンタの点は、グラフェンから寄与された電子状態を示します。 f グラフェン/ Cu(111)の総電荷密度と、top-fccジオメトリの独立したグラフェンとクリーンなCu(111)表面の電荷密度の合計との間の電荷差。等高線の間隔は0.004eÅ -3 です。
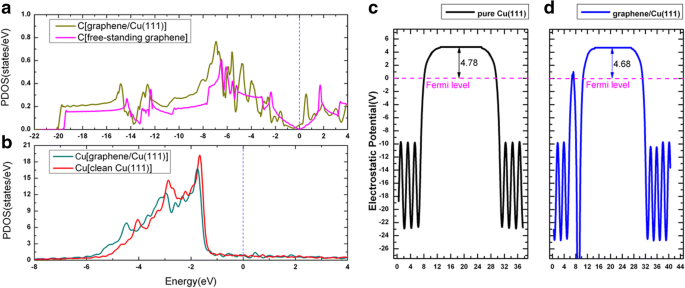
a のグラフェン/ Cu(111)の予測状態密度 C原子と b Cu原子。 c、d きれいなCu(111)表面とグラフェン/ Cu(111)界面の仕事関数
グラフェンで覆われたCu(111)表面へのバナジウムの吸着
このセクションでは、グラフェンで覆われたCu(111)表面へのバナジウム吸着の原子的、電子的、および磁気的特性を評価しようとします。図1dに示すように、topA、bridge、topB、およびhollowサイトとしてラベル付けされた4つの可能な吸着サイトが考慮されます。 1 / 9MLおよび1MLでのGra / Cu(111)上のV原子の吸着エネルギーをそれぞれ図1e、fに示します。グラフェンで覆われたCu(111)表面へのV吸着にエネルギー的に有利なサイトは、カバレッジに依存します。より具体的には、V吸着原子は1/9 MLに対して最大に調整された中空サイト(図1dを参照)を好みますが、1 MLの高いカバレッジには低調整されたトップサイト(つまり、topAサイト、図1dを参照)が優先されます。 。吸着エネルギー、V原子とその隣接C原子間の結合長、およびV / Gra / Cu(111)システムのバナジウムと炭素の原子磁気モーメントを表1に示します。 Gra / Cu(111)表面、E ad は、1 / 9MLおよび1MLのV原子あたり1.91および1.16eVであり、Cu(111)表面のものと比較するとある程度減少しています。明らかに、グラフェンバッファ層の導入は、予想どおり、V吸着原子とCu(111)表面の間の相互作用を弱める可能性があります。さらに、さまざまなカバレッジでのV / Gra / Cu(111)のVの強磁性次数のスピン偏極エネルギーを調査します。スピン偏極エネルギーは1 / 9MLでは390meVですが、1MLではスピン偏極はありません。スピン偏極エネルギーは、V / Cu(111)システムと比較した場合にV / Gra / Cu(111)システムでかなり高くなります(110meVと比較して390meV、表1を参照)。 1 / 9MLでのV / Gra / Cu(111)システムのV吸着原子の磁気モーメントは2.93μ B これは3μ B に近いです / atom(気相V原子の値)。これは、V原子が十分に分離されており、V原子とグラフェン層の間で電荷がほとんど移動しないことを意味します。 C原子の小さな磁気モーメント(0.16μ B / atom)も見つかります。
次に、グラフェンで覆われたCu(111)表面へのV吸着のバンド構造について説明します。図8は、Gra / Cu(111)表面へのVの吸着のバンド構造と、2つの異なるユニットセルにプロットされた自立グラフェンのバンド構造を示しています。その中で、青と赤の円は、それぞれグラフェンとV吸着原子からの寄与を表しています。まず、フェルミ準位を横切るCuのバンドが多数あることに注意してください。これは、システム内のCu原子がシステムのコンダクタンスに大きく寄与することを示しています。 1/9 MLカバレッジの場合、バンド構造(図8b、cを参照)は、システムの電子構造がスピン偏極していることも示しています。前のセクションで述べたように、グラフェンとCu(111)表面の間の相互作用は、Gra / Cu(111)では非常に弱いです。したがって、グラフェンに起因するバンドは、バンド構造全体で容易に認識されます。ただし、グラフェンで覆われたCu(111)表面(つまり、V / Gra / Cu(111)システム)へのV吸着後、グラフェンのディラック点が完全に「破壊」されていることがわかります(図8bを参照)。スピンアップチャネルのバンド構造、グラフェンの「線形交差点」はスピンダウンチャネルでも区別できます。それにもかかわらず、スピンダウン成分の「線形交差点」の非常に大きなダウンシフトは、グラフェン層への比較的多数の電荷移動を示しています。グラフェン層に移動する電荷は、C層とCu層の間の相互作用が弱いため、V原子の層から発生することに注意してください(図10aを参照)。 1/9 MLのスピンアップチャネルでは、大量の d を除いて 基板のCu原子の電子はフェルミ面に寄与し、 d もたくさんあります V吸着原子の電子はシステムのコンダクタンスに寄与します。一方、 p のハイブリダイゼーション 両方の d を持つC原子の電子 V吸着原子と表面Cu原子の電子が見えます(ただし、目立たない)。同様に、図8bのフェルミ面上の2つの代表的な点(A、B)にラベルを付けました。ポイントAは、 d からの貢献を表します xy 、 d x 2 − y 2 V吸着原子の電子、点Bは p の混成軌道からの寄与を示しています z d の両方を持つC原子の電子 z 2 V原子の電子とCu原子の最上層。明らかに、表面のV層は重要な導電層です。対照的に、1/9 MLのスピンダウンチャネルでは、フェルミ準位の電子状態は主に d から発生します。 Cu原子と p の電子 z C原子の電子; d の貢献 V吸着原子の電子はごくわずかです。 1 MLのカバレッジの場合、図8e、fに示すバンド構造は、システムがスピン偏極していないことを意味します。グラフェン層のディラック点も「破壊」されるため、V吸着原子とグラフェンバッファ層の間の相互作用は強くなるはずです。システムの計算された電子状態から、フェルミ準位に寄与する電子は主に s からのものであることがわかります。 、 d Cuと d の電子 V原子の電子と p -C原子の電子。より詳細には、フェルミ面にあるk点、A、B、C、D、およびEに図8eのラベルが付けられています。ポイントAは、 d のハイブリダイゼーションを示します yz および d z 2 最初の層(最上部)のCu原子の。ポイントB、C、およびDは、 d からの電子状態です。 -V吸着原子の電子。より具体的には、点Bは d からの電子状態を表します xy 、 d x 2 − y 2 V吸着原子の電子。さらに、ポイントEは s の強い混成を示しています 、 d z 2 p のV吸着原子の電子 z p の比較的弱い混合とともにC原子の電子 z d のC原子の電子 z 2 Cu原子の最上層の電子。

Band structures of a free-standing graphene, plotted in the Brillouin zones of a a (3 × 3) unit cell and d a (1 × 1) unit cell. Band structures of V adsorptions on the graphene-covered Cu(111) surfaces for b spin up of 1/9 ML, c spin down of 1/9 ML, e spin up of 1 ML, and f spin down of 1 ML
We now present the density of states for the V/Gra/Cu(111) system. The total density of states of V adsorption on the graphene-covered Cu(111) surface, together with the projected densities of states, are demonstrated in Fig. 9 for both the 1/9 ML and 1 ML coverages. At 1/9 ML, the spin polarizations of V adatoms and C atoms in the graphene layer are clearly seen (see Fig. 9c, d), while no spin polarization is found for Cu atom (see Fig. 9b). At 1 ML coverage, no spin polarization has been found for all atoms (see Fig. 9f-h). At 1/9 ML coverage, the DOS of the spin-up channel at the Fermi level is mainly contributed from the Cu atoms (totally 11.9 states/eV∙u.c.) and V atoms (totally 5.8 states/eV∙u.c.), with only minor contributions from the graphene layer (totally 0.4 states/eV∙u.c.). Meanwhile, the DOS of spin-down channel at the Fermi level is mainly contributed from the Cu atoms (totally 11.9 states/eV∙u.c.) and graphene layer (totally 1.1 states/eV∙u.c.), with only minor contributions from the V atoms (totally 0.1 states/eV∙u.c.). For the 1 ML coverage, both the DOS of spin-up and spin-down channels at the Fermi level are mainly contributed from the Cu atoms and V atoms (i.e., 1.1 and 0.7 states/eV∙u.c. for each spin component, respectively), with negligible contribution from the graphene layer (0.04 states/eV∙u.c). By integrating the PDOSs for each atom before and after the V adsorptions (leading to number of electrons), the charge transfer can be determined for different atoms. To be specific, the total valence electrons of the Cu atoms are reduced slightly for both the 1/9 ML and 1 ML coverages when compared with those of a clean Cu(111) surface, while the total valence electrons of C atoms are slightly increased when compared with that of a free-standing graphene. This implies that small amount of charges are transferred from Cu substrate to graphene layer for V/graphene/Cu(111) systems regardless of the V coverages. The total valence electrons of Cu atoms in V/Gra/Cu(111) systems are almost equal to those in the graphene/Cu(111) systems, which indicates that the Cu substrate has not been affected by V adsorption. The physical pictures given by the analysis of DOSs here are all in consistent with the analysis of the band structures. Finally, we show in Fig. 10 the contour plots of the deformation charge densities for the 1/9 ML and 1 ML coverages, respectively. The deformation charge density is defined as \( \Delta \rho \left(\overrightarrow{r}\right)={\rho}_{\left[V/ Gra/ Cu(111)\right]}\left(\overrightarrow{r}\right)-\sum \limits_{\mu =1}^N{\rho}^{atom}\left(\overrightarrow{r}-\overrightarrow{R_{\mu }}\right) \). As shown in Fig. 10, the interactions between the graphene layers and the substrate Cu atoms are both relatively weak for 1/9 ML and 1 ML coverages, which are in consistent with the above discussions. From Fig. 10a, for the 1/9 ML, the bonding between V adatoms and its adjacent C atoms is mainly ionic, and the covalent bonding is not obvious. In contrast, for the 1 ML coverage, both ionic and covalent bonding between V adatom and its adjacent C atoms are clearly visible (see Fig. 10b). Besides, the covalent bonding between neighboring V adatoms is also very significant at 1 ML coverage. Due to the existence of graphene buffer layer, V adatoms cannot interact directly with the Cu atoms.

a TDOS of V adsorption on graphene-covered Cu(111) surface at 1/9 ML coverage; PDOS for b Cu atoms, c C atoms, and d V adatoms at 1/9 ML. Likewise, e TDOS of V adsorption on Gra/Cu(111) surface at 1 ML coverage; PDOS for f Cu atoms, g C atoms, and h V adatoms at 1 ML. For comparison, PDOS of Cu atoms of a clean Cu(111) and graphene/Cu(111) are implanted in b and f , while PDOS of C atoms of a free-standing graphene are also implanted in c and g

Deformation charge densities for the adsorption of V atoms on the graphene-covered Cu(111) surface at two coverages, i.e., a 1/9 ML and b 1 ML. Electron accumulation and depletion are represented by magenta solid lines and green dashed lines, respectively. The green, black, and red balls represent Cu, C, and V atoms, respectively
We have also calculated the phonon spectra for both the V/Cu(111) and V/Gra/Cu(111) systems. From the calculated phonon spectra, we find that there is no “imaginary frequency” for both the two types of systems, indicating that the systems studied are dynamically stable and would be seen in the laboratory. Since the main purpose of our work is not the thermodynamic stability, therefore the figures of the phonon dispersions are not shown in this text. Second, we have noticed that the different DFT functionals we adopted may lead to the different results. Hence, we have calculated the 1 ML V/Gra/Cu(111) system (as a representative) within the DFT framework under the B3LYP, HSE06 hybrid functionals, as well as the PBE functional. The results suggest that the adsorption site with largest adsorption energy is the topA site, calculated from all the PBE, HSE06, and B3LYP methods. However, relative adsorption energies at different adsorption sites from the B3LYP and PBE and HSE06 methods differ significantly (results from PBE and HSE06 methods are almost the same, since this is a metallic system). On the other hand, the geometrical parameters obtained from the three functionals show good consistency. Although the detailed charge density contours are somewhat different between PBE and B3LYP method, the main bonding characteristics are the same from both the two methods. In summary, the main point is that the adsorption energies obtained from B3LYP functional are significantly larger than those from the PBE and HSE06 functionals. To explain this point, Paier et al. argued that B3LYP functional lacked of a proper description of the “free-electron-like” systems with a significant itinerant character (e.g., metals and small gap semiconductors). They have concluded that the overestimation of the total energy of the atoms can be induced by the significantly overestimation on the exchange and correlation energies of B3LYP functional. In this respect, PBE functional often shows much more reliable results [55].
結論
To summarize, using first-principles calculations, we have systematically investigated the electronic and geometric properties of the adsorption of V atoms on both the clean Cu(111) surface and the graphene-covered Cu(111) surface. Firstly, for the V/Cu(111) system, an adsorption site underneath the Cu surface layer is found as the preferable adsorption site for V atom regardless of the coverages. The hybridization of V’s d states with Cu’s d states rules the electronic properties of V/Cu(111) systems. Ferromagnetic order of V adatoms is energetically favored for 1/9 ML coverage (1.34 μB /atom), while no magnetism of V adatoms is observed for 1 ML coverage. Due to the strong interaction between V adatom and its adjacent substrate’s Cu atoms, the magnetic moment of V is significantly reduced. Secondly, the graphene/Cu(111) systems are investigated and the results agree well with the previous literatures. Thirdly, adsorptions of V on the graphene-covered Cu(111) at two coverages (i.e., 1/9 ML and 1 ML) show different preference of adsorption sites. The hollow site with maximum coordination is energetically favored for the adsorption of 1/9 ML, while the top site with low coordination is preferred for 1 ML adsorption. In V/Gra/Cu(111) systems, the interactions of C atoms with the V adatoms destroy the electronic properties of both the original graphene layer and the adsorbed atoms, represented by the strong hybridization of C’s p z -states with V adatoms’ d z 2 -states. A net magnetic moment for C atoms of graphene also appeared (0.16 μB/per carbon). In short, our study paves the way to a deep understanding of the adsorption properties of vanadium atoms on the clean Cu(111) and graphene-covered Cu(111) substrates. Simultaneously, this study also provides a reference for the possible applications of the V/Cu(111) and V/Gra/Cu(111) systems in the catalyst in nanomaterials industry, spintronic devices, and others.
略語
- Cu(111):
-
(111) surface of copper
- FM:
-
Ferromagnetic
- GGA:
-
Generalized gradient approximation
- ML:
-
Monolayer
- PAW:
-
Projector augmented wave
- PBE:
-
Perdew-Burke-Ernzerhof
- V/Cu(111):
-
Vanadium atoms adsorbed on Cu(111) surface
- V/Gra/Cu(111):
-
Vanadium atoms adsorbed on graphene-covered Cu(111) surface
- VASP:
-
Vienna ab initio simulation package
- vdWs:
-
van der Waals interactions
ナノマテリアル
- コバルトをドープしたFeMn2O4スピネルナノ粒子の調製と磁気特性
- TiO2ナノ流体に向けて—パート1:準備と特性
- 遷移金属をドープしたカオリナイトナノクレイの構造と電子特性
- 垂直電場によるML-GaSの電子的および光学的異方性特性の変調
- フェニルトリメトキシシランで修飾されたアルミナナノ粒子をベースにしたAl2O3:SiOCナノコンポジットの形成と発光特性
- 界面層の設計によるZnO膜の表面形態と特性の調整
- フェムト秒レーザー誘起硫黄ハイパードープシリコンN + / Pフォトダイオードの光学的および電子的特性
- Ag n V(n =1–12)クラスターの構造的、電子的、および磁気的特性の調査
- InSeナノリボンの電子構造とI-V特性
- Pr2CuO4ナノシートの制御された合成と選択的吸着特性:メカニズムの議論
- 自動車 PCB の特性と設計上の考慮事項