


黒フォスフォレンへの遷移金属の吸着:第一原理研究
要約
黒フォスフォレンは、独自の特性と幅広い用途を持つ新しい二次元材料です。第一原理計算を使用して、12種類の遷移金属(TM; Fe、Co、Ni、Cu、Ru、Rh、Pd、Ag、Os、Ir、Pt、およびAu)のフォスフォレンへの吸着挙動を調査しました。我々の結果は、すべての吸着システムが大きな結合エネルギーを持っていることを示しました。 Fe-、Co-、およびAu-フォスフォレンシステムは、磁気モーメントが2、1、および0.96 μの磁気状態を示します。 B それぞれ、これは、これらのシステムが磁性半導体であることを意味します。 TM-フォスフォレンへの酸素分子の吸着も調べた。興味深いことに、すべてのO 2 -(TM-フォスフォレン)システム(O 2 を除く) -(Pd-フォスフォレン)は、O–O結合を伸ばすことができます。これは、COの酸化における触媒としての用途に不可欠です。また、O 2 分子はO 2 を可能にします -(Fe-、Ni-、Cu-、Ir-、Rh-、Ag-、およびAu-フォスフォレン)システムが磁性半導体になり、O 2 -(コフォスフォレン)ハーフメタリック状態を表示します。私たちの結果は、フォスフォレンベースの触媒作用とスピントロニクスに重要な影響を与えると予想されます。
背景
しわの寄ったハニカム構造に配置されたリン原子の単分子層であるフォスフォレン[1,2,3]は、直接半導体性[4]、室温での超高移動度[4,5,6]、優れた機械的柔軟性などの独自の特性を備えています。 [7]、および高い熱電性能[8,9,10]。これらの特性により、フォスフォレンは電界効果トランジスタ[1、11、12、13、14、15、16]、リチウムおよびNaイオン電池[17、18、19]などのさまざまなアプリケーションに非常に適した材料になります。太陽電池[20、21]、光触媒[22]、スピントロニクス[23]、およびガスセンサー[24、25、26]。ただし、フォスフォレンは非磁性材料であり、その用途を広げるためにいくつかの戦略を採用する必要があります。
二次元(2D)材料の場合、吸着は通常、特定のアプリケーションの磁性を誘発するアプローチとして選択されます。以前は、Cao etal。 [27]は、グラフェンの電子的および磁気的特性が、Fe、Co、Ni、およびCuの吸着原子によって効果的に変調できることを示しました。 Kaloni etal。 [28]は、第一原理計算を使用して、Ti、V、Cr、Mn、Fe、およびCoで装飾されたシリセンシステムに磁気モーメントを誘導できることを示しました。 Ersan etal。 [29]は b -アルセネンは、H、B、C、P、Ge、As、およびSb原子の吸着後にスピン偏極特性を示しました。さらに、 w -アルセネンは、H、B、N、P、Cl、Ti、As、およびSbの吸着原子で正味の磁気モーメントを達成できます。黒フォスフォレンについては、Kulish etal。 [30]は、Ag-、Au-、Ti-、V-、Cr-、Mn-、Fe-、およびCo-フォスフォレンはかなり安定しており、理論計算ではさまざまな範囲の磁気モーメントを誘導できると予測しました。さらに、フォスフォレンにさまざまな原子を吸着させることで、さまざまな種類の電荷キャリアの特性を調整することもできます。 Ding and Wang [31]は、第一原理計算を使用して、フォスフォレンに吸着された原子の構造的、電子的、および磁気的特性を体系的に示しました。彼らは、吸着原子がフォスフォレンに磁性を導入し、P、Co、およびAu吸着原子が安定した磁気特性を誘導する可能性があることを指摘しました。 HuとHong [32]は、第一原理計算を使用して、フォスフォレン上の金属吸着原子の磁気特性を実証しました。彼らは、フォスフォレンの表面にCr、Fe、Co、またはAu原子を吸着させることにより、フォスフォレンに磁性が得られることを示しました。さらに、彼らは、Fe-フォスフォレン吸着システムが有望な希薄磁性半導体材料になると予測しました。したがって、黒色フォスフォレンへの遷移金属(TM)の吸着は、材料の磁気特性を効果的に調整することが期待できます。
上記の調査は、黒色フォスフォレンへの遷移金属の吸着挙動を研究しましたが、いくつかの問題は未解決のままです。たとえば、以前の研究では、主にフォスフォレンに吸着された3dTMの特性に焦点が当てられていました。 4dおよび5dTMは、フォスフォレンの特性をどのように設計しますか?また、フォスフォレンに吸収された貴金属も単原子触媒として利用できます。 Li etal。 [33]は、Auを吸着したシリセンは、COの酸化に対する触媒エネルギー障壁が低い高活性触媒である可能性があることを示唆しています。フォスフォレンに吸収された貴金属もCOの酸化の良い候補になるでしょうか。これらの質問に答えるために、この論文では、黒色フォスフォレンに吸着された12種類の遷移金属原子の構造的、磁気的、および電子的特性に関する詳細な第一原理研究の結果を示します。バルク相の強磁性金属である元素Fe、Co、およびNiを選択しました。反磁性である元素Cu; COの酸化に非常に効果的な貴金属Ru、Rh、Pd、Ag、Os、Ir、Pt、Au [19、34,35,36,37,38,39,40,41,42 、43、44、45]。フォスフォレンは12の金属すべてと強い結合を形成し、すべてのTM-フォスフォレンシステムはかなり堅牢であることがわかりました。フォスフォレンの電子的および磁気的特性は、吸着原子によって効果的に調整できます。さらに、ほとんどのTM-フォスフォレン吸着システムがCOの酸化における触媒の良い候補であることもわかりました。この調査の結果は、フォスフォレンの基礎研究に使用でき、多くの重要な分野での潜在的な用途を広げることもできます。 。
メソッド/実験
私たちの計算はスピン偏極密度汎関数理論(DFT)に基づいており、Vienna Ab Initio Simulation Package(VASP)[46、47]とPerdew-Burke-Ernzerhof( PBE)機能的[48,49,50]。 Grimme [51]のDFT-D3法を使用して、ファンデルワールス相互作用を計算しました。平面波基底関数系で400eVのエネルギーカットオフが採用されました。計算では、総エネルギーが1×10 -5 に収束するまで原子を緩和しました。 eVであり、各原子の残留力は0.01 eV /Å未満でした。ジグザグとアームチェアの方向に沿った大きなスーパーセル(4×3)を使用して、隣接するユニットセル間の相互作用を回避しました。格子定数は a に設定されました =13.20Åおよび b =13.74Å。 z に20Åの真空空間を適用しました 隣接する中間層間の相互作用を最小限に抑える方向。最適化中、Monkhorst-Pack [52] k -3×3×1のポイントグリッドを採用し、 k -7×7×1のポイントグリッドが総エネルギー計算に使用されました。
結果と考察
最初に、元のフォスフォレンの構造特性を調べました。図1aは、結晶構造の上面図と側面図を示しています。フォスフォレン単分子層は2つの原子面で構成されており、フォスフォレンのユニットセルは4つのP原子で構成されていることがわかります。フォスフォレン単分子層は、平衡格子定数 a の正方晶格子を持っています。 =3.30Åおよび b =4.58Å。水平方向のP–P結合の長さ( l 1 )は2.22Åですが、反対方向の長さ( l 2 )は2.26Åです。自然のままのフォスフォレンの直接バンドギャップは0.89eVであり(図1b)、伝導帯の最小値(CBM)と価電子帯の最大値(VBM)の両方がГポイントにあります。得られた格子定数とバンドギャップは、以前の調査研究[30,31,32,53]で得られた値と非常に一致しています。
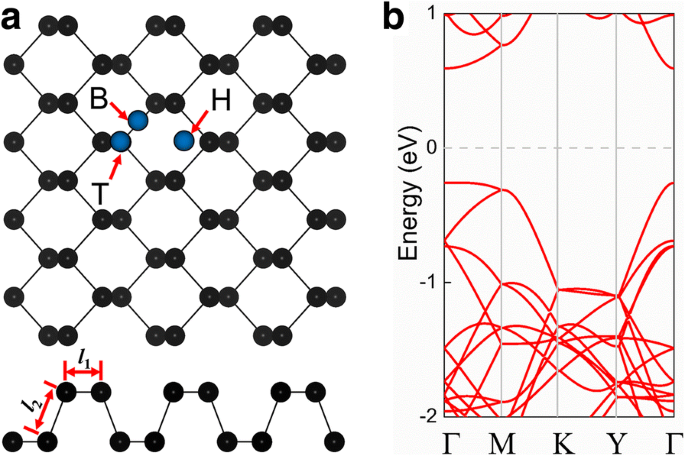
a 自然のままのフォスフォレン(4×3×1スーパーセル)の結晶構造の上面図と側面図。青い円は、中空スポット(H)、2つのリン原子間のブリッジ(B)、およびリン原子(T)の上部に吸着された不純物原子の典型的な位置を表しています。 b 純粋なフォスフォレンの電子バンド構造と最初のブリルアンゾーン。フェルミ準位はゼロに設定されています
典型的な吸着原子は、常に3つの位置のいずれかに吸着されます。中空サイトの上(H)、2つのリン原子間のブリッジ(B)、およびリン原子の上(T)です。フォスフォレンへの吸着原子の吸着エネルギーを計算し、次の関係を使用して吸着システムの安定性を調べました。
$$ {E} _ {\ mathrm {ad}} =\ left({E} _ {\ mathrm {TM}} + {E} _ {\ mathrm {phosphorene}} \ right)-{E} _ {\ mathrm {TM}-\ mathrm {フォスフォレン}} $$(1)ここで E TM は孤立した金属原子のエネルギー、 E フォスフォレン は元のフォスフォレン層の総エネルギーであり、 E TM-フォスフォレン は吸着システムの総エネルギーです。この式に基づくと、吸着エネルギーが大きいほど、構造が安定していることを示します。私たちの研究で研究されたすべての金属原子は、フォスフォレンのHサイトに留まることを好むことがわかりました。表1に示す、フォスフォレンのHサイトに吸着された金属原子の計算された吸着エネルギーは、2〜6eVの範囲で変化します。 TM-フォスフォレンの結合長( d TM-P )は短く、2.11〜2.43Åの範囲であることが実証されました。より悪い電荷分析[54,55,56]は、0.16、0.16、0.07、0.17、0.32、0.33、および0.16 | e |を示しています。 (4d-TM)-フォスフォレンおよび(5d-TM)-フォスフォレン吸着システムでは、Ru、Rh、Pd、Os、Ir、Pt、およびAuの金属原子からそれぞれフォスフォレンに移動します。これらの結果はすべて、TM吸着原子とフォスフォレンの間の化学結合の形成を示しています。さらに、これらの結果は最近の研究[30,31,32]に近いものです。
<図>表1に示すように、Ni-、Cu-、Ru-、Rh-、Pd-、Ag-、Os-、Ir-、およびPt-フォスフォレンシステムは非磁性状態を示しますが、Fe-、Co-、およびAuは非磁性状態を示します。 -フォスフォレンシステムの磁気モーメントは2、1、および0.96 μです。 B 、 それぞれ。スピン偏極電荷密度(ρ =ρ スピンアップ − ρ スピンダウン )も図2に示され、磁性TM-フォスフォレン吸着システムの磁性の起源と分布を調べています。これらの各場合の磁気モーメントは主に吸着原子から発生し、小さな磁気モーメントは最近傍から発生します。さらに、Fe-、Co-、およびAu-フォスフォレンシステムの計算されたバンド構造を図2に示します。これらのシステムはすべて、それぞれ0.38、0.22、および0.06eVのバンドギャップを持つ磁性半導体であることがわかります。スピントロニックアプリケーションに役立ちます。
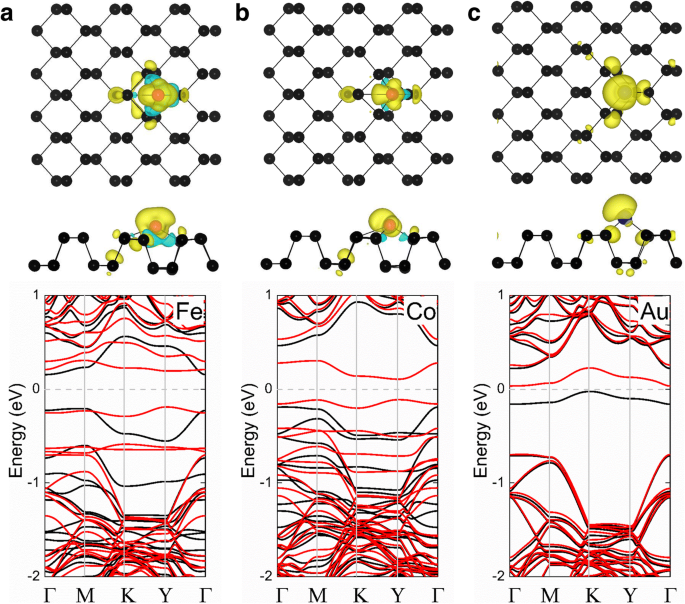
a のスピン密度 Fe-フォスフォレン、 b コフォスフォレン、および c Au-フォスフォレンシステムが一番上の行に示されています。各システムの対応するバンド構造が下の行に示されています。黒と赤の球は、それぞれP原子とTM原子を表しています。上の行は、電荷密度の等表面値が0.002 e /Å 3 のスピン偏極電荷密度のプロットです。 各TM-フォスフォレンシステムの元のフォスフォレンの結晶構造の上面図と側面図に重ねて表示されます。黄色とシアンの領域は、それぞれアップスピンとダウンスピンに対応しています。バンド構造のプロット(下の行)では、黒と赤の線はそれぞれスピンアップチャネルとスピンダウンチャネルを示しています。フェルミ準位はゼロに設定されており、灰色の破線で示されています
次に、O 2 の吸着挙動を調べました。 TM-フォスフォレン系のTM原子の上に。 O 2 の吸着のための2つの典型的なエネルギー最低構成 TM-フォスフォレンシステム(O 2 -(TM-フォスフォレン))を図3に示します。O 2 の場合 -(Fe-フォスフォレン)、O 2 -(コフォスフォレン)、O 2 -(Cu-フォスフォレン)、O 2 -(Pd-フォスフォレン)、およびO 2 -(Pt-フォスフォレン)システム、O 2 分子はフォスフォレンのジグザグ方向に平行であり(図3a)、O–P結合長はそれぞれ1.84Å、1.86Å、2.04Å、2.18Å、2.05Åです。 O 2 の場合 -(Ni-フォスフォレン)、O 2 -(Ru-フォスフォレン)、O 2 -(Rh-フォスフォレン)、O 2 -(Ag-フォスフォレン)、O 2 -(Os-フォスフォレン)、O 2 -(Ir-フォスフォレン)、およびO 2 -(Au-ホスホレン)システムでは、分子は表面から特定の角度で、フォスフォレンのジグザグ方向に沿っています(図3b)。一方、TM吸着原子の周りの2つの隣接するO原子は同等ではありません。結果を表2に示します。吸着エネルギー( E ad )of O 2 O 2 で -(TM-フォスフォレン)システムは次のように計算されました:
$$ {E} _ {\ mathrm {ad}} ={E} _ {\ mathrm {TM}-\ mathrm {phosphorene}} + {E} _ {{\ mathrm {O}} ^ 2}-{E } _ {{\ mathrm {O}} ^ 2- \ mathrm {TM}-\ mathrm {フォスフォレン}} $$(2)ここで、\({E} _ {{\ mathrm {O}} ^ 2- \ mathrm {TM}-\ mathrm {phosphorene}} \)、 E TM-フォスフォレン 、および\({E} _ {{\ mathrm {O}} ^ 2} \)は、O 2 の総エネルギーです。 -(TM-フォスフォレン)システム、TM-フォスフォレンシステム、およびO 2 それぞれ分子。表2に示すように、O 2 の吸着エネルギーは2.659、1.850、0.970、0.906、2.402、1.548、0.001、0.786、3.109、1.980、0.416、1.029eVです。 -(Fe-フォスフォレン)、O 2 -(コフォスフォレン)、O 2 -(Ni-フォスフォレン)、O 2 -(Cu-フォスフォレン)、O 2 -(Ru-フォスフォレン)、O 2 -(Rh-フォスフォレン)、O 2 -(Pd-フォスフォレン)、O 2 -(Ag-フォスフォレン)、O 2 -(Os-フォスフォレン)、O 2 -(Ir-フォスフォレン)、O 2 -(Pt-フォスフォレン)、およびO 2 -(Au-フォスフォレン)システム、それぞれ。すべての場合において、O 2 の吸着エネルギーを除いて大きな吸着エネルギー -(Pd-フォスフォレン)システムは、O 2 化学吸着されます。
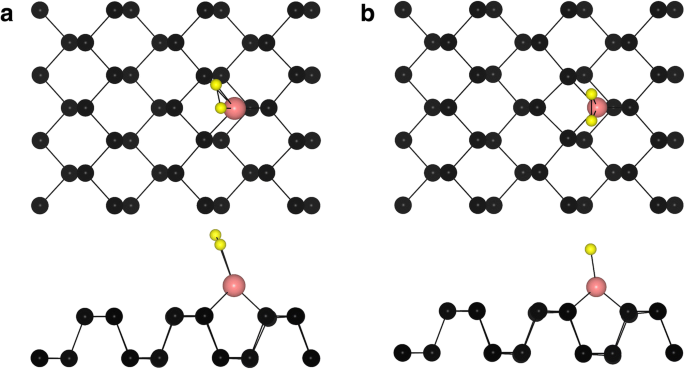
O 2 の典型的な吸着サイトの上面図と側面図 TM-フォスフォレン上の分子。黒、ピンク、黄色の球は、それぞれP、TM、O原子を表しています
O–O結合の伸長は、COの酸化における触媒のLangmuir-HinshelwoodメカニズムとEley-Ridealメカニズムの両方にとって重要であることがかなり認識されています[57]。一般的に、O–O結合長が長いほど、触媒反応が容易になります。各システムのO–OおよびTM–O結合長も表2に示されています。明らかに、O–O結合は元のO 2 の1.23Åから増加しています。 おそらくO 2 が原因で、吸着分子の分子がそれぞれ1.38、1.36、1.32、1.35、1.40、1.34、1.32、1.30、1.46、1.39、1.40、1.32Åになります。 電子受容体です。さらに、ほとんどのO 2 のTM–Oの結合長 -(TM-フォスフォレン)システムは、O 2 間の相互作用のために短いです とTM原子。この結合長は1.84〜2.19Åの範囲で変化し、化学結合が形成されます。特に、吸着されたO 2 では、O–O結合が1.40Åに伸長します。これはシステムの中で最も高い値です。 Pt-フォスフォレン系の分子。したがって、Pt-フォスフォレンシステムは、おそらく高い触媒能力を持っているため、COの酸化の触媒として非常に適しています。
これらのシステムの高活動の根本的なメカニズムについてより多くの洞察を得るために、O 2 を選択しました。 -例として(Pt-フォスフォレン)を使用し、その局所状態密度(LDOS)を調査しました。図4aは、Pt-フォスフォレンシステムのPtのd軌道、O 2 のPtのd軌道に投影されたLDOSを示しています。 -(Pt-フォスフォレン)システム、O 2 のO–O結合 -(Pt-フォスフォレン)システム、および気相O 2 。図4aの上部パネルでは、 E に1つのピークが見られます。 F − 0.6 eV。これは、Pt-フォスフォレン系のPtの部分的に占有されたd軌道に由来します。これらの状態は、Pt-フォスフォレンシステムの高い活性に関与しているはずです。 O 2 の吸着後 分子、フェルミ準位より下のPtのd軌道に投影されたLDOSは、O 2 の吸着後にシフトダウンされます。 電荷移動による分子であり、フェルミ準位より上の状態も大幅に増加します。一方、LDOSは吸着されたO 2 に投影されました 分子は、O 2 2 π * 軌道(最低空軌道、LUMO)は部分的に占有されており、ガス値 E からシフトダウンしています。 F + 2eVから E F − 0.1eV。明確にするために、O 2 の電荷密度の差 -(Pt-フォスフォレン)システムも紹介されています。
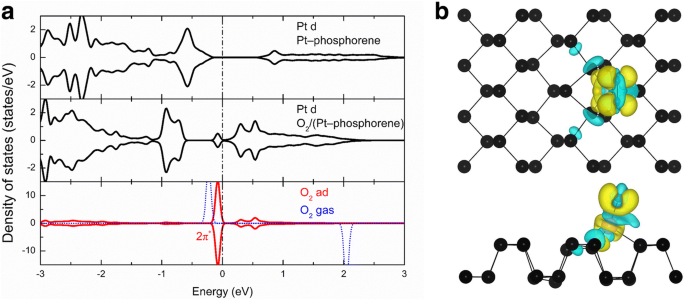
a PtおよびO 2 の局所状態密度(LDOS) Pt-フォスフォレンとO 2 の分子 -Pt-フォスフォレンシステムと気相O 2 、 それぞれ。 b O 2 の電荷密度の違い -(Pt-フォスフォレン)システム;黄色の領域(つまり、+ 0.002e /Å 3 )およびシアン領域(つまり、-0.002e /Å 3 )電子密度の増加と損失にそれぞれ対応します
電荷密度の差は次のように定義されます。
$$ {\ varDelta} _ {\ rho} ={\ rho} _T-{\ rho} _ {\ mathrm {molecule}}-{\ rho} _ {\ mathrm {absorbed}} $$(3)ここで、ρ T 、ρ 分子 、およびρ 吸収 O 2 の合計料金です -(Pt-フォスフォレン)システム、O 2 それぞれ、分子、およびPt-フォスフォレンシステム。図4bに示すように、O 2 に局在する大きな黄色の領域 分子は、Pt-フォスフォレンからO 2 への有意な電子移動があることを示しています 、これは、O 2 間の強い軌道混成も示しています。 およびPt-フォスフォレンシステム。 Bader電荷分析[54,55,56]によると、0.19 | e | Pt-フォスフォレン系からO 2 に移動します 分子。したがって、大きな電荷移動はO 2 の反結合性状態を満たします。 分子であり、O–O結合を大幅に弱めます。同様に、他のシステムの高活性の根本的なメカニズムは、O 2 間の電荷移動によっても理解できます。 分子とTM-フォスフォレンシステム。実際、より悪い電荷分析[54,55,56]は、-0.68、-0.50、-0.42、-0.52、-0.46、-0.24、-0.24、-0.37、-0.53、-0.25、-0.19、および− 0.09 | e | TM-フォスフォレンからO 2 の酸素分子に移動します -(Fe-フォスフォレン)、O 2 -(コフォスフォレン)、O 2 -(Ni-フォスフォレン)、O 2 -(Cu-フォスフォレン)、O 2 -(Ru-フォスフォレン)、O 2 -(Rh-フォスフォレン)、O 2 -(Pd-フォスフォレン)、O 2 -(Ag-フォスフォレン)、O 2 -(Os-フォスフォレン)、O 2 -(Ir-フォスフォレン)、O 2 -(Pt-フォスフォレン)、およびO 2 -(Au-フォスフォレン)システム、それぞれ。
最後に、O 2 の磁気特性を調べました。 -(TM-フォスフォレン)システム。 O 2 の磁気モーメント -(TM-フォスフォレン)システムを表3に示します。O 2 -(Ni-フォスフォレン)、O 2 -(Cu-フォスフォレン)、O 2 -(Rh-フォスフォレン)、O 2 -(Ag-フォスフォレン)、およびO 2 -(Ir-フォスフォレン)システムの磁気モーメントは2.00、1.00、1.00、1.14、および1.00 μです。 B それぞれ、常磁性O 2 の吸着に起因します。 分子。これらのO 2 のスピン偏極電荷密度 -(TM-フォスフォレン)システムを図5に示します。O 2 の場合 -(Fe-フォスフォレン)およびO 2 -(コフォスフォレン)システムでは、磁気モーメントは主に遷移金属原子とO 2 から発生すると考えられています。 分子。それどころか、O 2 の場合 -(Ni-フォスフォレン)、O 2 -(Cu-フォスフォレン)、O 2 -(Rh-フォスフォレン)、O 2 -(Ag-フォスフォレン)、O 2 -(Ir-フォスフォレン)、およびO 2 -(Au-フォスフォレン)システム、磁気モーメントは主にO 2 から発生します 分子。これらの仮説は、表3に表示されている結果と一致しています。ガス分子の吸着がO 2 の電子構造にどのように影響するかをよりよく理解するため -(TM-フォスフォレン)システム、各システムの電子バンド構造を計算し、その結果を図5に示します。最初に、フェルミ準位( E )の周りにフラットバンドが発生することを発見しました。 F )O 2 の吸着後 主にO 2 からのすべてのシステムの分子 分子。 O 2 の場合 -(Fe-フォスフォレン)、O 2 -(コフォスフォレン)、O 2 -(Ni-フォスフォレン)、O 2 -(Cu-フォスフォレン)、O 2 -(Rh-フォスフォレン)、O 2 -(Ir-フォスフォレン)、O 2 -(Ag-フォスフォレン)、およびO 2 -(Au-フォスフォレン)システム、スピンアップおよびスピンダウンスプリットのチャネルは磁気特性を明らかにします。 O 2 -(Fe-フォスフォレン)、O 2 -(Ni-フォスフォレン)、O 2 -(Cu-フォスフォレン)、O 2 -(Ir-フォスフォレン)、O 2 -(Rh-フォスフォレン)、O 2 -(Ag-フォスフォレン)、およびO 2 -(Au-フォスフォレン)は磁気半導体挙動を示し、O 2 を除いてかなりのバンドギャップがあります。 -(Co-ホスホレン)システム。半金属であることが明らかになりました。これらの結果は、このシステムがフォスフォレンベースのスピントロニクスに応用できる可能性があることを示唆しています。
<図>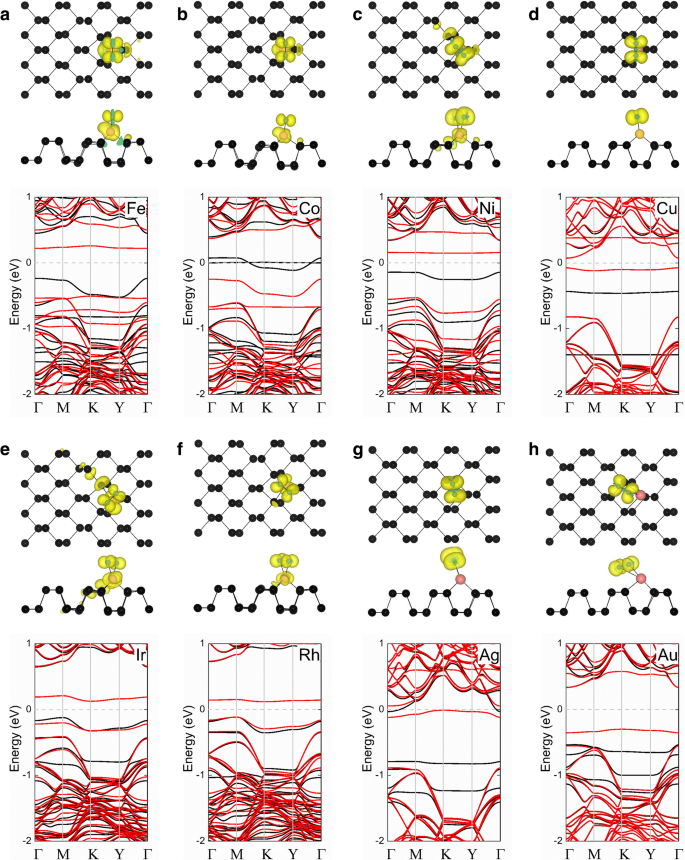
a のスピン密度 O 2 -(Fe-フォスフォレン)、 b O 2 -(コフォスフォレン)、 c O 2 -(Ni-フォスフォレン)、 d O 2 -(Cu-フォスフォレン)、 e O 2 -(Ir-フォスフォレン)、 f O 2 -(Rh-フォスフォレン)、 g O 2 -(Ag-フォスフォレン)、および h O 2 -(Au-フォスフォレン)システムが一番上の行に表示されます。各システムの対応するバンド構造が下の行に示されています。上の行は、電荷密度の等表面値が0.002 e /Å 3 のスピン偏極電荷密度のプロットです。 純粋なフォスフォレンの結晶構造の上面図と側面図に重ねて表示されます。黄色とシアンの領域は、それぞれアップスピンとダウンスピンに対応します。バンド構造のプロットでは、黒と赤の線はそれぞれスピンアップチャネルとスピンダウンチャネルを示しています。フェルミ準位はゼロに設定されており、灰色の破線で示されています
結論
さまざまなTM-フォスフォレンシステムの構造的、電子的、および磁気的特性を調査しました。すべての吸着原子は、フォスフォレンの中空サイトを占めることを好むことがわかりました。かなりの吸着エネルギーは、すべてのTM-フォスフォレン吸着システムがかなり堅牢であることを示しており、フォスフォレンが12種類すべてのTM吸着原子と強い結合を形成していることを示しています。さらに、Fe、Co、およびAuをドープすると、単層フォスフォレンに磁気半導体特性が生じ、総磁気モーメントが2、1、および0.96 μになることがわかりました。 B それぞれ。
さらに、O 2 の特性も調べました。 TM-フォスフォレン系に吸着した分子。すべてのO 2 を見つけることは非常に励みになりました -(TM-フォスフォレン)システム(O 2 を除く) -(Pd-フォスフォレン)、O–O結合の伸長により、COの酸化に対して優れた触媒活性を示します。 O 2 -(Fe-フォスフォレン)、O 2 -(Ni-フォスフォレン)、O 2 -(Cu-フォスフォレン)、O 2 -(Rh-フォスフォレン)、O 2 -(Ag-フォスフォレン)、O 2 -(Ir-フォスフォレン)、およびO 2 -(Au-フォスフォレン)システムは、磁気モーメントが2.00、2.00、1.00、1.00、1.14、1.00、および1.00 μのスピン偏極半導体特性を示します。 B 。 O 2 -(コフォスフォレン)は、磁気モーメントが2.00 μの磁気ハーフメタリック特性を示します。 B 。したがって、私たちの結果は、触媒作用とスピントロニクスの分野でフォスフォレンを適用するための新しい可能性を開く可能性があります。
略語
- 2D:
-
二次元
- B:
-
ブリッジ
- GGA:
-
一般化された勾配近似
- H:
-
中空サイト
- LDOS:
-
局所状態密度
- PBE:
-
Perdew-Burke-Ernzerh
- T:
-
リン原子の上部
- TM:
-
遷移金属
ナノマテリアル