


遷移金属をドープしたカオリナイトナノクレイの構造と電子特性
要約
この作業では、一連の遷移金属(Cr、Mn、Fe、およびCo)をドープしたカオリナイトナノクレイを密度汎関数理論(DFT)計算によって調査しました。カオリナイトの幾何学的構造と電子構造に及ぼす金属ドーピングの影響を分析した。遷移金属(TM)をドープしたカオリナイト構造の強磁性(FM)、反強磁性(AFM)、および非磁性(NM)状態を調べました。結晶体積、格子定数、結合長、電荷、およびスピンは、分散補正密度汎関数理論(DFT-D2)によって計算されました。結果は、Cr 3+ およびFe 3+ ドーパントはAFM状態でより安定しているのに対し、Mn 3+ AFMとFMの両方の状態を優先し、Co 3+ ドーパントの好ましいNM状態。また、遷移金属のドーピングは、バンドギャップに格子体積の膨張といくつかのドーパント状態を引き起こす可能性があります。
背景
カオリングループのナノクレイ鉱物は、熱水変質および/または風化プロセスの結果として、層状構造、小さな粒子サイズ、そして最も重要なことに、ヒドロキシル基が豊富な水和表面のために、独特の物理的特性を持っています。材料化学、環境化学、鉱物物理学の研究者の注目を集めています[1,2,3,4,5,6,7,8,9,10,11]。地球上で最も豊富なナノクレイ鉱物の1つであるカオリナイトは、プラスチック、触媒作用、およびセメント産業で広く使用されています。新規担体材料としてのカオリナイトのさらなる機能化は、様々な分野でますます注目を集めている。カオリナイトは、他のナノ粒子と混合して太陽エネルギーユーティリティ用の相変化材料を形成するための支持材料として機能するか[4、5]、またはドープされた酸化物でコーティングして導電性分野での用途向けの導電性粉末を形成することができます[9、12]。カオリナイトと機能性ナノ粒子の混成は、相乗効果により、Pd–ZnOの光触媒活性とCdSの発光特性を向上させることがわかりました[6、7]。カオリナイトの表面特性は、いくつかの官能基を表面に固定することによって[13、14]、またはさらなる改善のために酸活性化前処理によって変更されました[2]。
カオリングループの鉱物の構造とエネルギーは、実験的に[15,16,17]、理論的に[18,19,20,21,22]広く研究されています。カオリナイト表面への重金属吸着の理論的研究は、Cd、Cu、Hg、およびNi(II)吸着について研究され[23]、イオンに対するカオリナイト粘土の吸着能力は、Ni> Cu> Cd>の順に見出されました。 Hg(II)。カオリナイト(001)表面へのPb(II)[24、25]とウラニル[26]の吸着と拡散が研究され[24、25、26]、水系での吸着挙動も後で報告されました[27、 28]。カオリナイト表面へのMg、Ca、Feのドーピングの影響、およびその後のH 2 の吸着と浸透 中間層へのOが研究された[29]。 H 2 の吸着エネルギー ドープされたカオリナイト(001)上のOは、ドープされていない表面よりも少ないことがわかりました。固有の欠陥がある場合とない場合のカオリナイトの電子構造は、標準密度汎関数理論(DFT)汎関数と混成汎関数によって研究されています[30]。しかし、最近まで、カオリナイトの脱ヒドロキシル化、脱アルミニウム、およびシリカ凝縮プロセス中の構造進化がDFT計算によってモデル化されていません[1、31、32]。カオリングループの材料からAlを除去すると、これらの層状材料の形状と電子特性が大幅に変化し、サポート効果が向上しました[1、2]。
化合物の構造と特性を変更するためのよく知られた方法としての金属ドーピングは、Al 2 について理論的に研究されてきました。 O 3 [33]、TiO 2 [34]、MOF [35]、およびその他の固体[36]。遷移金属(TM)ドーピングによるカオリナイトナノクレイの構造と特性の変化を調査することは、この層状粘土材料にとって興味深いことです。この作業では、一連のCr、Mn、Fe、およびCoをドープしたカオリナイトナノクレイをDFT計算によって研究し、カオリナイトナノクレイの幾何学的構造と電子構造に対する金属ドーピングの影響に焦点を当てました。これらの遷移金属をドープしたカオリナイト構造の可能な強磁性(FM)、反強磁性(AFM)、および非磁性(NM)状態を研究しました。格子定数、結合長、電荷、およびスピンは、分散補正密度汎関数理論(DFT-D2)によって最適化および計算されました。
メソッド
すべての計算は、第一原理DFTに基づいたプログラムCASTEP(Cambridge Sequential Total Energy Package)コード[37]を使用して実行されました。計算には、Perdew、Burke、およびErnzerhof(PBE)による交換相関ポテンシャルを使用した一般化勾配近似(GGA)が使用されました[38]。ファンデルワールス分散相互作用を説明するために、GrimmeのDFT-D2分散補正が含まれています[39]。ウルトラソフト擬ポテンシャル平面波形式を使用して、500eVのエネルギーカットオフが適用されました[40]。 Monkhorst–Pack [41]グリッド、2×2×3 k -点メッシュは、幾何学的緩和と電子構造計算に使用されました。基底状態での自己無撞着な全エネルギーは、密度混合スキームによって効果的に得られました[42]。ジオメトリの最適化では、自己無撞着場(SCF)許容誤差の収束しきい値を1.0×10 -6 に設定しました。 eV /原子、原子にかかるすべての力は0.03 eV /Å未満に収束し、総応力テンソルは0.05 GPaのオーダーに減少し、最大イオン変位は0.001Å以内でした。原子価状態で調査された元素はO(2s 2 2p 4 )、Al(3s 2 3p 1 )、Cr(3s 2 3p 6 3d 5 4秒 1 )、Mn(3d 5 4秒 2 )、Fe(3d 6 4秒 2 )、およびCo(3d 7 4秒 2 )。 Mn、Fe、CoにはUspcc擬ポテンシャルを使用し、残りの元素にはusp擬ポテンシャルを使用しました。 Broyden–Fletcher–Goldfarb–Shanno(BFGS)最小化アルゴリズムを使用したジオメトリ最適化中に、セルパラメータと原子協調が完全に緩和されました。結晶の対称性は、電子基底状態がより低い対称性を採用できるように、TMイオンに異なる初期磁気モーメントを課すことによって削除されました。
結果と考察
初期のカオリナイト構造は、以前の研究[1]で採用されました。図1は、カオリナイト(4カオリナイトユニット)の緩和された2×2×1結晶構造を示しています。カオリナイト層構造、Al 2 Si 2 O 5 (OH) 4 は、八面体のAl–Oシートと四面体のSi–Oシートで構成され、頂端のO原子(O a )。 Si–O四面体は、1つの中央のSi原子と4つの周囲のO原子で構成され、そのうちの1つはO a です。 原子と他の3つは基底O原子(O b )。 Al–O八面体は、中央の1つのAlと周囲の6つのOで構成され、そのうち2つはO a です。 原子と他の4つは、他のAl–O八面体と共有されるO原子(OHグループ内)です。さらに、これらのOH基は2種類に分けることができます:層間OH(OH inter )層構造の表面と内部OH(OH 内部 )AlシートとSiシートの間の層構造の内側。したがって、Si–O結合には、Si–O a の2種類があります。 およびSi–O b (黒い点線)、および3種類のAl–O結合、Al–O inter (赤い点線)、Al–O 内側 (緑の点線)、およびカオリナイトバルク構造のAl–O(黒の点線)。
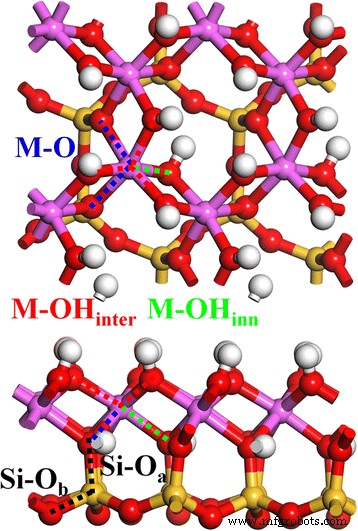
トップ(アップ )とサイド(ダウン )カオリナイトの眺め。 Si–O a (黒 )、Si–O b (黒 )、M–OH inter (赤 )、M–OH 内部 (緑 )、およびM–O(青 )結合は点線で示されます
分散エネルギーは、層間の相互作用により、粘土鉱物の構造安定化において常に主要な役割を果たします[21、43]。いくつかの混成汎関数の中で、PBE-D2 [21]、B3LYP [22]、B3LYP-D [18]、およびRPBE-D2 [18、21]は、カオリナイトの実験的な格子構造を取得するために使用されました[44、45 ]、PBE-D2汎関数は、正確で時間のかからないことがわかりました。以前に簡単に報告されたように、結合長に対するPBE汎関数の過大評価は、実験結果と比較した分散補正によって克服されます[1]。カオリナイトの構造に対するTMドーピングの影響を区別するために、ここではまず、格子構造と、中心陽イオン(SiおよびAl)と酸素原子間の最適化された結合距離O a を再検討します。 、O b 、およびOH inn 。
表1に示すように、カオリナイトの場合、分散補正されたPBE-D2汎関数を使用して最適化された計算された単位格子体積は実験値に近く、PBE汎関数(〜3.4%)と比較して大幅に低い相対誤差(〜0.4%)が得られます。 。格子ベクトルaおよびbの場合、PBE-D2を使用した相対誤差(〜0.4%)は、PBE(〜1.1%)よりもはるかに低くなります。また、PBE-D2の分散補正では、カオリナイトの層距離(ベクトルc)が0.17Å(約2%)減少します。特に、分散補正後の格子角は、特にαの場合、実験結果に非常に近いです。カオリナイトの結合長分布については、PBE-D2ではSi–O a の改善はほとんど見られません。 、Al–OH 内部 、および実験結果と比較したAl–O結合では、Al–OH inter が大幅に改善されています。 Al–O表面での結合(表面化学にとって重要)とSi–O b のわずかな改善 Si–O表面で結合します。特に、Al–OH inter の場合 結合、PBE-D2からの分散補正は、Al–O表面の最外層での結合環境を正確に表しているようです。これは、上にある別のカオリナイト層のSi–O表面からの分散力の影響を強く受けます。ここで言及するもう1つのポイントは、実際には2つの分割されたAl–O結合(図1、青い点線)があり、結合長が約1.95Åと2.00Åと大きく異なることです[45]。これはAl–Oの格子歪みを示しています。八面体は、Si–OシートとAl–Oシートの格子不整合に起因します。実験結果と比較したカオリナイト構造の計算における主要なエラーとして、これらのAl–O結合は、同様の平均結合長を持つPBEとPBE-D2の両方によって過大評価されています(表1)。 PBE-D2は、約1.96Åと2.04Åの2つのAl–O結合を与え、2番目の結合は0.04Å過大評価されています(図2、青い点線)。
<図>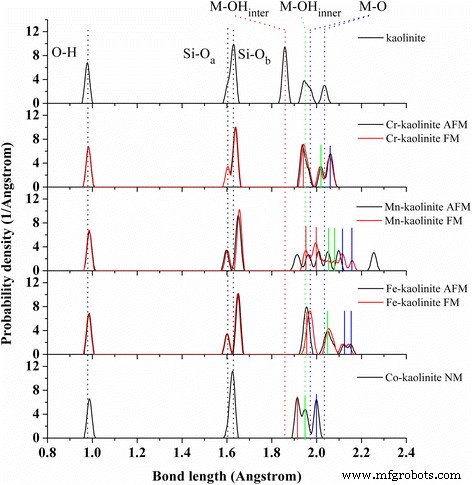
Cr–、Mn–、Fe–、およびCo–カオリナイトの結合分布。 TM-カオリナイトごとに多磁性状態が示されています。平均化されたさまざまなタイプのO–H(黒 )、Si–O a (黒 )、Si–O b (黒 )、M–OH inter (赤 )、M–OH 内部 (緑 )、およびM–O(青 )カオリナイトの結合は点線で示されます 。 M–OH inter (赤 )、M–OH 内部 (緑 )、およびM–O(青 )Cr-カオリナイトの結合( AFM )、Mn-カオリナイト( FM )、Fe-カオリナイト( AFM )、およびコカオリナイト( NM )は実線で示されます
遷移金属(Cr、Mn、Fe、およびCo)をドープしたカオリナイトは、Al原子をCr、Mn、Fe、またはCo原子に置き換えることによって構築されました。 Al 3+ の同等の置換のみ TM 3+ のイオン +3以外の化学状態でTMイオンを非等価に置換すると、電荷バランスのために追加の空孔または不純物が生じるため、イオンが考慮されました。構造の観点から、TM-カオリナイトのPBEおよびPBE-D2汎関数は、カオリナイトで観察されたのと同様の構造の違いを示します。 PBE-D2汎関数は、カオリナイトの2つの基底面の格子ベクトルと結合長についてより適切に記述されることを考慮すると、TM-カオリナイトに関する議論に続いて、主にPBE-D2汎関数によって得られた結果に依存しました。 TMをドープしたカオリナイトの格子定数、結合長、電荷、スピン、およびそれらの磁気状態を表1にまとめました。Cr-カオリナイト、Mn-カオリナイト、およびFe-のAFM状態とFM状態のエネルギー差(TM原子あたり)カオリナイトはそれぞれ0.022、-0.006、0.094eVです。コカオリナイト構造は非磁性状態でのみ安定しているため、コカオリナイトのNM構造のみを示しています。
TM-カオリナイトの単位格子体積はカオリナイトと比較して拡大しており、Mn-カオリナイト> Fe-カオリナイト>> Cr-カオリナイト>>カオリナイト> Co-カオリナイトの傾向があります。セルの拡張は、主にAl–O結合と比較して長いM–O結合によって引き起こされ、格子ベクトルaおよびbの主要な拡張につながります。一方、Si–O b Si–Oシートの結合は同時に伸長し、それに応じてαとβの結晶格子角が歪んでいます。 FM状態のMn-カオリナイトのセル体積はAFM状態と比較して1.4%増加しますが、対照的に、Cr-カオリナイトとFe-カオリナイトのセル体積に対する磁気秩序の影響はほとんど見られません。 Cr、Mn、Fe、Coの磁気モーメントは、TMをドープしたAl 2 の磁気モーメントに近い。 O 3 [33]、マリケン電荷はわずかに高く、これはより強い反応性を意味します。
TM-カオリナイトの結合長分布を図2に分析します。TM-カオリナイトのさまざまなタイプのSi-OおよびM-O結合は、各ドーピング元素の実線で示されています。全体的に言えば、M–OとSi–O b の結合長が増加しています。 TMドーピング後、その間、M–OH inter の分割されたM–O結合の結合分布が再編成されます。 (赤)、M–OH 内部 (緑)、およびM–O(青)結合。特に、分割されたAl–O結合(青い点線)は、CrとCoのドーピング後に消失しました。さらに、結合長の分布は、Mn原子の磁気秩序に大きく依存しますが、CrおよびFe原子の影響はわずかです。
Cr 3+ のPDOS結果 (d3)、Mn 3+ (d4)、Fe 3+ (d5)、およびCo 3+ (d6)および対応する電荷密度分布を図1および2に示します。ヤーン・テラーの定理によれば、縮退した電子システムは、周囲の結合環境の影響を受ける縮退[46]を取り除くように、自然に歪みます[47]。 TM 3+ の場合 たくさんのヒドロキシル基を持つカオリナイトの八面体Alサイトへのドーピング、TM 3+ の5つのd殻軌道 トリプレットt 2g に分割されます 状態とダブレットe g 正八面体の状態。三重項状態の電子は、配位子間の中間領域に局在し、最も近いO状態とさらにハイブリダイズします。二重項状態にあるものは、配位子を直接指しているため、t 2g よりもエネルギーが高くなります。 電子。一般に、e g 内の電子の存在 軌道は八面体結合を不安定にする傾向があり、縮退は、満たされた軌道の反対側の結合を長くし、空の軌道の反対側の結合を短くすることによって除去されます。 TM 3+ のd–d遷移 (ああ)種は常に占領されたt 2g からのものです 占有されていないe g への軌道(dxy、dyz、およびdzx) 軌道(d x2-y2 またはd z2 、占有率に応じて)。 e g 間の軌道分割 軌道とt 2g Cr 3+ の軌道 (d 3 )、Mn 3+ (d 4 )、Fe 3+ (d 5 )、およびCo 3+ (d 6 )TM-カオリナイトのそれはAl 2 のそれと類似しています O 3 およびTiO 2 [33、48、49]が、3d軌道間の分裂エネルギーは、おそらく周囲のヒドロキシル基との混成のために、それ自体の酸化物よりもわずかに大きくなっています(図3)。
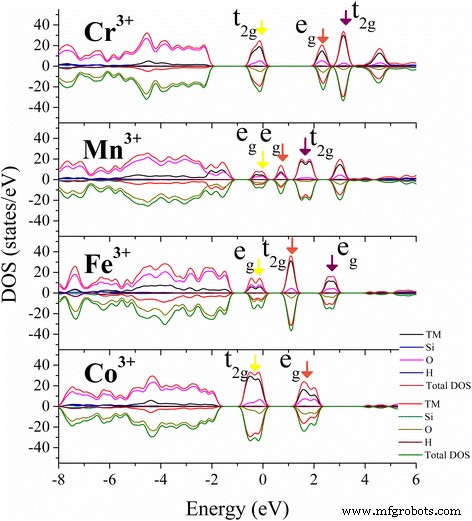
TMドープカオリナイトの最も安定した状態の総状態密度(DOS)と原子投影状態密度(PDOS)が示されています。最も占有されている3D軌道(黄色 )と最初の(茶色 )と2番目(紫 )フェルミ準位の周りの最も低い空いている3D軌道は、色付きの矢印で示されます。
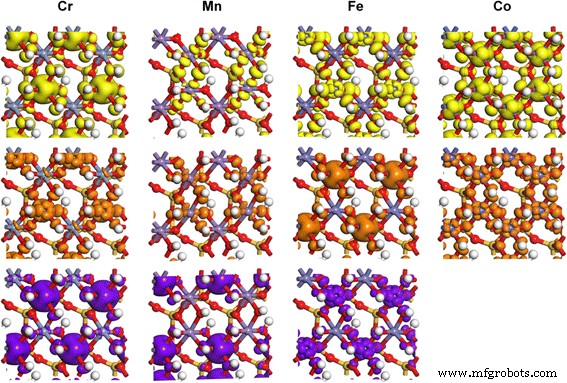
矢印で示される状態に対応する、TM-カオリナイトのTM3d軌道の部分電荷密度 PDOSの結果で。等値面レベルは0.02e /Å 3
Mn-カオリナイトのFM状態とAFM状態の分裂エネルギーの差は小さく、状態密度の分布はスピン方向が異なることを除いて類似しています。したがって、簡単にするために、AFM状態の結果のみが示されています。高スピンMn 3+ の場合 (d 4 )AFM状態のMn-カオリナイト中のイオン、2つのe g のうちの1つのみ 軌道は価電子帯の最大値(VBM)で占められています(図3、黄色の矢印)。 d z2 の職業 エネルギーが低い軌道は、 z に沿った2つの配位子の結合電子に強い反発力を与えます 軸を作成し、M–O結合をその方向に延長します。この効果は、よく知られているヤーン・テラー効果です。伝導帯最小値(CBM)の下部の状態は、最も低い空いているd z2 で構成されます。 軌道(茶色の矢印)とより高いd x2y2 Mn 3+ の軌道(紫色の矢印) (d 4 )。 Cr 3+ の場合 (d 3 )、Fe 3+ (d 5 )、およびCo 3+ (d 6 )ドープされた場合、t 2g およびe g 軌道は均等に占有されており、ヤーン・テラー歪み効果の影響は小さく、TM-カオリナイトのM-O結合のわずかな偏差しか生じませんでした(図2)。 TMドーピングによる構造と電子特性のそのような変更は、触媒作用[50、51]、CO捕捉[52、53]、薬物負荷[54]、およびエネルギー貯蔵[55,56,57]の分野でのカオリンの適用を改善する可能性があります。 ]。また、モンモリロナイト[50、58]、パーライト[55]、タルク[59]などの他の鉱物にも適用して、電子特性を変えることができます。
結論
カオリナイトナノクレイの幾何学的構造と電子構造に及ぼす遷移金属(Cr、Mn、Fe、Co)のドーピングの影響をDFT計算で調べます。結晶の体積、格子定数、結合長、電荷とスピン、および可能な磁気状態が計算され、研究されます。 Cr 3+ およびFe 3+ ドーパントは、AFM状態、Mn 3+ の下でより安定します。 FM状態を優先し、Co 3+ ドーパントはNM状態を好みます。遷移金属のドーピングは、格子体積の拡大とM–O結合分布の再編成を引き起こします。一方、TMドーパントは、カオリナイトのバンドギャップに大きな分裂エネルギーを持ついくつかの3d状態を導入します。
略語
- AFM:
-
反強磁性
- BFGS:
-
Broyden–Fletcher–Goldfarb–Shanno
- CASTEP:
-
ケンブリッジシーケンシャルトータルエネルギーパッケージ
- CBM:
-
伝導帯の最小値
- DFT:
-
密度汎関数理論
- DFT-D2:
-
分散補正密度汎関数理論
- FM:
-
強磁性
- GGA:
-
一般化された勾配近似
- NM:
-
非磁性
- PBE:
-
Perdew、Burke、Ernzerhof
- SCF:
-
自己無撞着フィールド
- TM:
-
遷移金属
- VBM:
-
価電子帯の最大値
ナノマテリアル
- スタックカップカーボンナノファイバーの原子および電子構造を明らかにする
- 垂直電場によるML-GaSの電子的および光学的異方性特性の変調
- ns-Laserによって調製されたブラックシリコン上に酸素をドープしたナノ結晶の電子状態と可視発光
- 微結晶およびナノセルロースの構造と誘電特性に及ぼす水の影響
- フェムト秒レーザー誘起硫黄ハイパードープシリコンN + / Pフォトダイオードの光学的および電子的特性
- Ag n V(n =1–12)クラスターの構造的、電子的、および磁気的特性の調査
- GeSiSnナノアイランドと歪み層を備えた半導体膜の形態、構造、および光学特性
- InSeナノリボンの電子構造とI-V特性
- 20種類の金属とその特性
- クロム金属:元素、特性、および用途
- 自動車 PCB の特性と設計上の考慮事項