


天然シルク繊維の酸化とナノ分散
要約
ナチュラルカイコ シルク(BS)と Antheraea pernyi シルク(AS)は次亜塩素酸ナトリウム(NaClO)溶液で酸化されました。その後、酸化シルクスラリーを超音波処理した後、個々のシルクナノファイバー(SN)が得られました。この場合、得られたSNの直径は約100 nm、長さは数マイクロメートルでした。薄い膜は、光学的に透明(75%以上の透過率)、機械的に堅牢(ヤング率の〜4.5 GPa)、および強化された濡れ特性を備えたSNをキャストすることによって形成されました。これらのSNを使用することによる興味深い凝集分散(再分散)プロセスは、pH値を調整することによって強く制御されました。その結果、負に帯電したSNは最大〜20 wt%(初期分散の100倍)に集中する可能性があり、保管、輸送、およびエンジニアリングアプリケーションに並外れたメリットをもたらします。
はじめに
階層構造を持つ材料は、自然の生物学的システムに遍在しています[1、2]。それらは、ポリマーの主要な特性と各階層での構造の機能的適応により、多様な機能を提供します[3,4,5]。このような特殊な特性を再現する機能を強化した人工材料を設計するために、ポリマーの元のナノ構造を保持する抽出プロセスが望まれています[6、7、8、9、10]。化学的、物理的、および生物学的アプローチを使用して、多糖類ナノファイバー(セルロースやキチンなど)を繊維複合構造から分離するためのさまざまな研究が行われています[11、12、13]。特に、2,2,6,6-テトラメチルピペリジン-1-オキシルラジカル(TEMPO)を介した天然セルロース/キチンの酸化とそれに続く穏やかな機械的処理を採用することにより、完全に個別化された高度に結晶性のナノファイバーが得られました[14、15 ]。しかし、経済的および環境的問題は依然として残っています。ナノフィブリル分離のためのこれらの既存の技術は、TEMPOやヘキサフルオロイソプロパノール(HFIP)などの高価なおよび/または有毒な試薬を必要とします。さらに重要なことに、得られたナノファイバー分散液の濃度が低いため、保管、輸送、および用途が制限されていました。
さまざまな昆虫やクモによって紡がれる動物の絹も、階層的な繊維構造を持っています[16、17]。これらのタンパク質分子は、ナノスケールからマクロスケールまで組み立てられたフィブリルの形をしており、絹材料に卓越した機械的および生化学的特性をもたらします[18、19、20、21]。ただし、シルクナノ構造を実現するには、(i)複雑な階層構造、(ii)高い結晶化度、および(iii)シルク繊維のミクロ/ナノフィブリル間の接着により、抽出プロセスが依然として課題となります。超音波処理は、絹繊維の分割に適用されています[22]。しかし、得られたナノファイバーは絡み合っており、加工性に欠けていました。塩-ギ酸システムを使用した絹繊維の部分的な溶解は、不安定な木のようなナノファイバーの束を示しました[23]。部分溶解と超音波処理を使用した統合アプローチにより、単一のナノフィブリルの直径に縮小された絹繊維が得られました[24]が、そのようなナノファイバーのアスペクト比と歩留まりはまだ改善されていません。
これらの問題に対処するために、フルサイズのメソシルクを抽出するための簡単でスケーラブルな戦略を練り上げました[25]。多糖類の単離[26]と同様に、カルボキシル基が Bombyx mori に導入されました。 シルク(BS)と Antheraea pernyi 静電反発力によって分散するナノファイバー用のシルク(AS)ファイバー。ただし、TEMPOや臭化ナトリウム(NaBr)などの冗長な化学物質は、選択的な酸化が不要であるため除外されました。ここでは、高アスペクト比の個々のナノファイバーを生成するためのこのプロセスの有効性を開示しました。得られたシルクナノファイバー(SN)膜では、光学的に透明で、機械的に堅牢で、濡れ性が向上しています。それらの多糖類ベースのナノファイバー(すなわち、セルロースおよびキチンナノファイバー)と比較して、SNの興味深い凝集-再分散特性はpH値によって調節されていました。
材料と方法
分解された絹繊維の酸化
分解された絹繊維は、生の Bombyx mori から調製されました。 (または Antheraea pernyi )カイコ繊維(Xiehe Silk Co.、中国)。簡単に説明すると、5 gの絹繊維を0.02M炭酸ナトリウム水溶液(重量比1:400)で30分間煮沸した後、蒸留水で十分に洗浄した後、風乾しました。次に、脱ガムした絹繊維を、重量比1:20のギ酸(88 wt%)溶液に浸しました。混合物を室温で少なくとも1時間インキュベートし、次に10,000 r / minで3分間ホモジナイズして、懸濁液を得た。懸濁液を8000r / minで遠心分離した後、分解された絹繊維が固体状態で得られました。
酸化のために、分解した絹繊維をpH 7に洗浄し、長さ数センチメートルの短い断片に切断し、必要な量の次亜塩素酸ナトリウム(NaClO)溶液を1gの分解した絹繊維とともに100mlの水に加えました。水酸化ナトリウム(NaOH)を混合物に継続的に添加して、pHを10に維持しました。NaOHの消費が観察されなくなったら、0.5 M塩酸(HCl)を滴下して、pHを7に調整することにより反応を停止しました。水不溶性画分を10000r / minで遠心分離し、数回洗浄しました。最後に、水不溶性画分を超音波ホモジナイザーで19.5 kHz、出力300 Wで20分間処理した後、シルクナノファイバーが得られました。長時間の超音波処理中の過熱を避けるために、氷水浴が採用されました。
酸化絹繊維のX線回折分析
X線回折(XRD)実験は、Cu-Kα源(λ)を備えたUltima IV多目的X線回折システム(Ultima IV、リガク、日本)を使用して実行されました。 =0.1542 nm)。 X線源の電圧と電流はそれぞれ40kVと30mAでした。酸化された絹繊維のデコンボリューションの結果は、PeakFitソフトウェア(4.0)を使用して分析されました。ピークの数と位置は、スペクトルからの2次導関数の結果から定義され、デコンボリューションプロセス中に固定されました。帯域幅はソフトウェアによって自動的に調整されました。
ナノファイバーの形態観察
さまざまなナノファイバーの形成を観察するために、分散液を0.01 wt%に希釈しました。走査型電子顕微鏡法では、希釈した分散液の10μLアリコートをシリコンウェーハ上に置き、風乾しました。サンプルを金とパラジウムでコーティングし、JEOL-JSM 7600F(JEOL、日本)SEMを使用して5kVの電圧で画像化しました。透過型電子顕微鏡(TEM)の場合、希釈された分散液の10μLアリコートをカーボンコーティングされたCu電子顕微鏡グリッドに配置しました。余分な液体は濾紙に吸収され、風乾されました。サンプルグリッドは、Titan 80-300(FEI、米国)透過型電子顕微鏡を使用して80kVで観察されました。ナノフィブリルのサイズは、米国の国立衛生研究所で開発されたImageJソフトウェア(1.48)を使用して分析されました。
機械的試験
BS、AS、CN(セルロースナノファイバー)、ChN(キチンナノファイバー)の膜は、溶媒蒸発法を使用して厚さ約50μmで鋳造されました。各ナノファイバーメンブレンは、長さ60〜80mm、直径5mmのいくつかのストリップに調整され、電子ユニバーサル試験機(AG-Xplus、島津製作所、日本)によって引き伸ばされて機械的特性が決定されました。このテストでは、フィクスチャの初期間隔は20mm、ストレッチ速度は1mm / minでした。
光学特性と濡れ特性
厚さ25μmのさまざまな膜の光透過率は、AmershamBiosciencesのUltrospec2100プロ分光計を使用して350〜800nmで測定されました。
接触角の測定には、ドロップメータ(協和インターフェースサイエンス株式会社)を採用しました。画像分析は、約0.5秒以内に膜に滴下された4μLの蒸留水滴の形状から自動的に実行されました。
結果と考察
シルクナノファイバーの酸化と分離
図1aは、絹繊維材料からナノファイバーを分離するための戦略を示しています。最初に前処理プロセスを使用して、ギ酸で処理することによりこれらの絹繊維を分解しました(追加ファイル1のラマンスペクトルに示されているように、アミノ酸またはギ酸とのヒドロキシル基の間で化学反応は発生しませんでした:図S1および追加の関連する議論ファイル)。この前処理により、絹繊維が5〜20μmの幅のマイクロファイバー構造に分解されました(図1a)。次に、次亜塩素酸ナトリウム(NaClO)を使用して、分解された絹繊維を酸化/部分的に溶解(分解)しました。 TEMPO(2、2、6、6-テトラメチルピペリジン-1-オキシラジカル)を介したTEMPOを使用した多糖類の酸化の条件に従って、水酸化ナトリウム(NaOH)を混合物に連続的に添加してpHを10に維持しました。 / NaClO / NaOHシステム。この場合、シルクフィブロインシーケンスの反応性アミノ酸が限られているため、TEMPOとNaBrはシルクファイバーの酸化に必要ありませんでした。初期の絹繊維は、タンパク質の〜0.3 mM / gのカルボキシル濃度を持っていました。これは、分子配列のアスパラギン酸とグルタミン酸に起因していました[27]。その後、酸化された絹のカルボキシル含有量は、セリン残基のヒドロキシメチル基の酸化のために、NaClOの添加後にほぼ直線的に増加した。 NaClOの添加量が20mM / gタンパク質に達したとき、酸化絹の最終的なカルボキシル濃度は、BSとASでそれぞれ0.889と1.013 mM / gタンパク質でした(図1b、c)。しかし、過剰な量のNaClOは絹繊維を劣化させた可能性があります[28]。たとえば、BSとASの水不溶性画分は、20 mM / gタンパク質のNaClO添加で、それぞれ58.52と69.30 wt%でした。酸化後の水不溶性画分の重量損失は、酸化中の限られた分解に関して、タンパク質の≤10mM / gのNaClO添加が許容可能であることを示唆しました(タンパク質の75%以上が残っていました)。したがって、タンパク質1グラムあたり10 mMのNaClOを使用して、BSおよびAS繊維を酸化しました。ここで、カルボン酸塩含有量は、BSおよびASでそれぞれ0.724および0.837 mg / gタンパク質です。
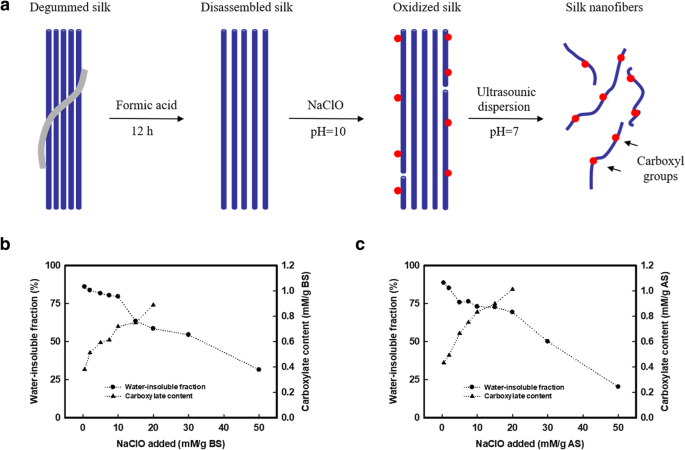
SNとBSおよびASのカルボキシル含有量のプロセス図。 a シルクファイバーのシルクナノファイバー(SN)への酸化と分散の概略図。 b カイコの酸化後の水不溶性画分のカルボキシル基含有量と残存重量 (BS)次亜塩素酸ナトリウム(NaClO)の添加に対応します。カルボキシル含有量は0.293から0.889mM / g BS(NaClOの添加は20 mM / gタンパク質)に増加し、58.52 wt%のタンパク質が残っていました。 c Antheraea pernyi の場合 シルク(AS)。カルボキシル含有量は0.347から1.013mM / gASに増加し(NaClOの添加は20 mM / gタンパク質でした)、69.30 wt%のタンパク質が残っています
最後に、水不溶性画分を超音波ホモジナイザーで処理した後、ナノファイバーが得られました(図2)。走査型電子顕微鏡観察により、酸化により絹がミクロレベルで緩み、直径数ミクロンの繊維が形成され、超音波処理によりさらに直径105±27 nmのナノ繊維に分散したことが明らかになりました(図2c)。絹繊維の表層をほとんど剥離する他のプロセス[24]と比較すると、酸化絹の静電反発力により、ナノファイバーの最終収率は酸化絹に基づいて約50%になります。同様の戦略がASファイバーにも適用されました。得られたASナノファイバーの直径は112±33nmで、輪郭の長さは1μmを超えていました(図2f)。
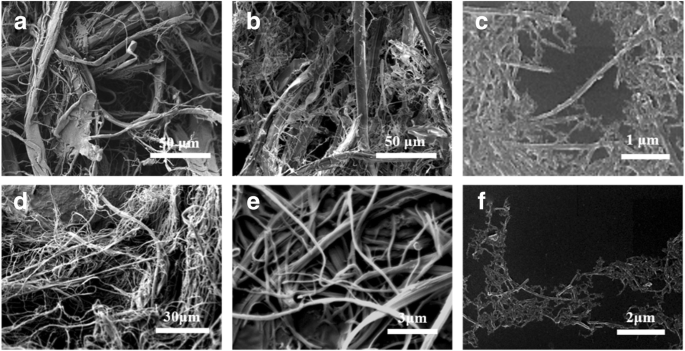
各プロセスで得られた絹繊維の代表的なSEM観察。 a ギ酸前処理後の分解されたBS繊維、 b 酸化されたBSファイバー、および c 直径105±27nmのBSナノファイバー。 d ギ酸前処理後の分解されたAS繊維、 e 酸化されたAS繊維、および f 直径112±33nmのASナノファイバー。 BSおよびASナノファイバーの輪郭の長さは1μm以上です
絹繊維の結晶化度
シルクタンパク質分子は、親水性-疎水性-親水性ポリマーとして機能し、疎水性コア(結晶領域)から伸びる親水性ビーズ(アモルファス領域)の形成中に不規則なサイズのミセルに折りたたまれました[17]。 SNは、ミセル間の外側領域の接着のために組み立てられました。しかし、絹繊維のNaClO酸化は、それらのナノ構造間の弱い接着を提案したことが示唆されています[25]。図3aおよびbに示すように、酸化後、酸化されたBSファイバーのX線回折(XRD)パターンは、元のパターン、および酸化されたASファイバーのXRDパターンと同様でした。したがって、酸化された絹繊維は、それらの天然のナノビルディングブロック、すなわち、絹繊維のβシート構造のままであった。一方、これらのXRDパターンのデコンボリューション(図3c、d)は、酸化後のBS繊維とAS繊維の両方で結晶化度の有意な変化を示唆しました。詳細は、表1に記載されています。ただし、酸化は主にセリン残基で発生しました。絹タンパク質の中で、NaClOによって攻撃される可能性のあるアモルファス領域にいくつかのアミノ基がありました[29]。したがって、表1の酸化されたBS繊維の結晶化度は、24.8%(分解されたBS)から41.3%(10 mM / gのタンパク質NaClOの添加)に増加し、続いてカルボキシル含有量が増加したことが理解できます。同様の傾向は、酸化されたAS繊維の場合にも見られ、これらのAS繊維の結晶化度は22.9%から39.2%に増加しました。結果は、静電反発力に加えて、絹タンパク質のアモルファス領域の破壊もSNを分散させる重要な要因であったことを示唆している。酸化された絹繊維(BSとASの両方)の結晶化度に続いて、NaClOの添加が<10 mM / gタンパク質の場合にカルボキシル含有量が増加しました。アモルファス領域の分解は、絹タンパク質の結晶化したコアの前にあります。ただし、過剰量のNaClO(20 mM / gタンパク質)は、シルクを劣化させる可能性があります。この現象は、図1bおよびcで明らかにした結果とよく一致しています。
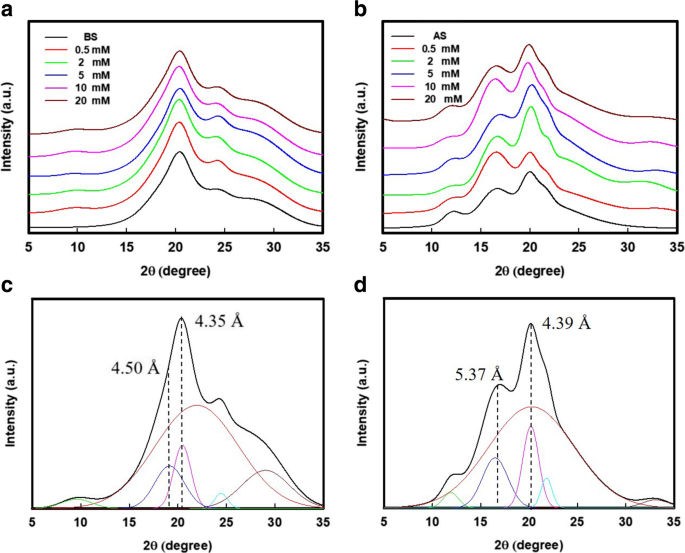
酸化された絹繊維のXRD分析。 a のX線回折(XRD)パターン BSと b 様々なNaClO添加で酸化したAS。 c の代表的なデコンボリューションと結果 BSと d ASマテリアル
シルクナノファイバーの性能
10mMのNaClO酸化絹繊維の超音波処理によって得られた酸化BSおよびASナノファイバーの形態を図4aおよびbに示します。 BSナノファイバーとASナノファイバーのアスペクト比は類似しており(ImageJソフトウェアで計算)、平均してBSナノファイバーが16.92、ASナノファイバーが19.12です。比較すると、TEMPOを介した酸化を使用して調製されたセルロースナノファイバー(CN)とキチンナノファイバー(ChN)を図1および2に示します。 4cおよびd。これらのSNをさらに特徴づけるために、溶媒蒸発法を使用して、約50μmの厚さの膜をキャストしました。光学的に透明な(75%以上の透過率)シルクメンブレンは、UV-Vis(350〜800 nm)分光光度計を使用して評価されました(図4e)。
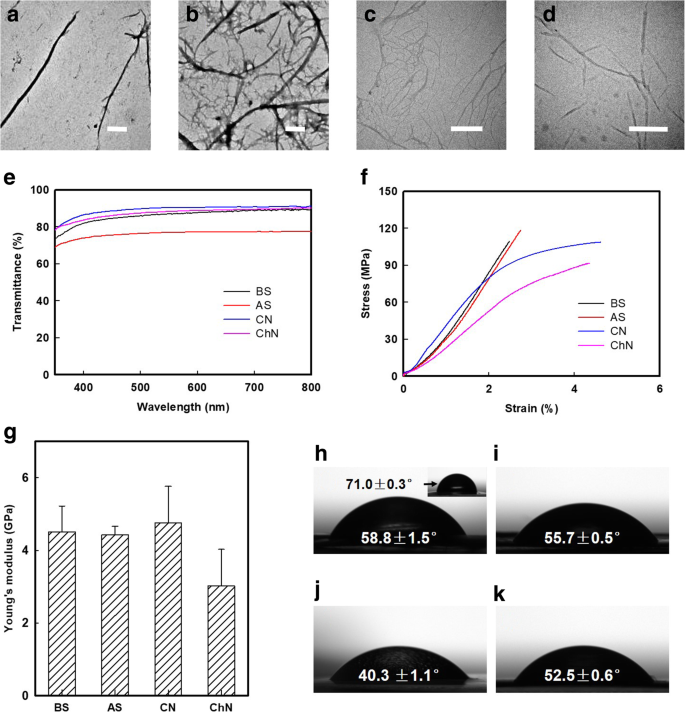
SN、CN、およびChNの形態および特性テスト。結果として生じる a の透過型電子顕微鏡(TEM)観察 BSと b 10 mM / gタンパク質NaClO添加により酸化したASナノファイバー、 c セルロースナノファイバー(CN)、および d TEMPOを介した酸化によって達成されたキチンナノファイバー(ChN)。スケールバーは500nmです。 e BS、AS、セルロース(CN)、およびキチン(ChN)ナノファイバーによってキャストされた約50μmの厚さの膜のUV-Vis透過率。 f BS、AS、CN、およびChNナノファイバーによってキャストされた約50μmの厚さの膜の代表的な応力-ひずみ曲線。 g BS、AS、CN、およびChNナノファイバーから鋳造されている膜のヤング率。データは平均SD( n =5)。 h – k f によってキャストされた膜の水接触角 BSナノファイバーは58.8±1.5°であり、再生されたBS膜のそれから大幅に減少しました(71.0±0.3°、挿入画像)。 AS、CN、およびChN膜では、それぞれ55.7±0.5、40.3±1.1、および52.5±0.6°の水接触角が示されました
このダウンサイジング法で得られたナノファイバーは、高い結晶構造と高いアスペクト比を維持していました。その結果、これらの膜は、BSおよびASでそれぞれ4.51±0.71および4.43±0.23 GPaのヤング率で堅牢な機械的特性(図4g)を示しました。これは、CNおよびChN膜(代表的なひずみおよび応力曲線を図4fに示します)。さらに、カルボキシル基の導入により、再生膜ではBS膜の濡れ性が大幅に向上しました。図4hに示すように、BSナノファイバー鋳造膜の水接触角は58.8±1.5°ですが、再生されたBS膜(図4hの挿入画像)は71.0±0.3°です。さらに、AS(図4i)、CN(図4j)、およびChN(図4k)膜では、それぞれ55.7±0.5、40.3±1.1、および52.5±0.6°の水接触角が示されました。
>CNとChNの両方とシルクデバイスは、同様の機械的堅牢性、加工可塑性、生化学的特性などにより、数十年にわたって材料科学に広く適用されてきました[13、30、31]。もちろん、これらの多糖類とタンパク質ベースの材料。したがって、それらの違いがナノファイバーの形成をどのように調節しているか疑問に思いました。適切に分散されたBSおよびAS分散液は、中性条件下で、それぞれ-39.5±0.66および-37.4±2.4mVのゼータ電位を示しました。カルボキシル基間の静電反発力は、シルクのマイクロ/ナノフィブリル界面間の接着に反します。したがって、これらのナノファイバーは水相に均一に分散しました。興味深いことに、pHが下がると、H + 図5aおよびbに示すように、負に帯電した表面をシールドして、ナノファイバーの凝集を引き起こしました。 SNの凝集体は、pH> 7に調整することで水中に再分散させることができます。または、遠心分離後に簡単に収集して、わずかに攪拌しながら再分散させることができます。図5の下のグラフは、さまざまなpH条件下で収集されたSN骨材の残りの重量を示しています。 BSの場合、凝集体の80.1±1.7および90.9±2.2 wt%(ASの場合は85.7±2.2および93.6±1.5 wt%)がpH5および3でそれぞれ回収されました。一方、このプロセスでは、SNを初期分散と比較して約100倍(〜20 wt%)濃縮し、濃度は〜0.2 wt%でした。 SNのこの魅力的な特性は、(i)タンパク質ベースの材料の固有のpH応答、および(ii)凝集および再分散プロセス中のソフトマターSNの柔軟性に起因していました。凝集-再分散現象は、これらのSNの薬物負荷および放出担体としての有望な用途を示唆しました。さらに、結果として得られるSNが保管および輸送に適していることに異論はありません。
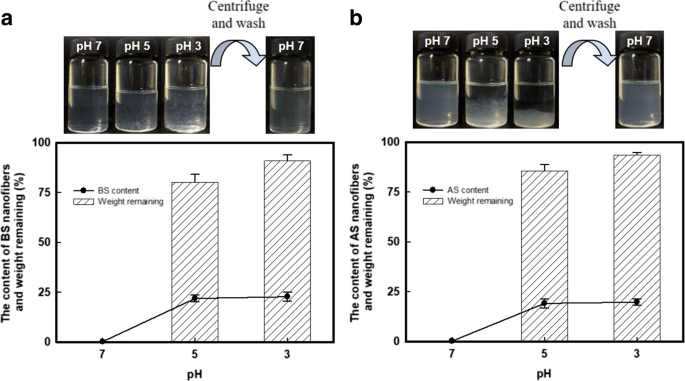
SNの再分散プロセス。 a のpH応答現象の写真 BSと b ASナノファイバー。遠心分離後、タンパク質の80 wt%以上(BSとASの両方)が残っており、タンパク質含有量は〜20 wt%
結論
要約すると、個々の分散したBSおよびASナノファイバーは、NaClO酸化後に達成されました。このアプローチは、ナノファイバーを調製するための多糖類のTEMPOを介した酸化と同様でした。ただし、TEMPO / NaBr触媒は必要ありませんでした。準備されたままのSNは、直径が約110 nm、長さが数ミクロンで、表面は負に帯電しています。 SN膜では、光学的に透明で、機械的に堅牢で、濡れ性が向上しています。特に、SNはpHを下げることによって〜20 wt%に濃縮することができ、これらのパルプ状のSNは中性水溶液に再分散可能でした。これらの結果に基づくと、SNは材料科学および生物医学アプリケーションの優れた候補です。
データと資料の可用性
この調査中に生成または分析されたすべてのデータは、この公開された記事に含まれています。
略語
- AS:
-
サクサン シルク
- BS:
-
カイコ シルク
- ChN:
-
キチンナノファイバー
- CN:
-
セルロースナノファイバー
- SEM:
-
走査型電子顕微鏡
- SN:
-
シルクナノファイバー
- TEM:
-
透過型電子顕微鏡
- XRD:
-
X線回折
ナノマテリアル