


長鎖ノンコーディングRNAMALAT1 / microRNA-143 / VEGFAシグナル軸は血管内皮損傷誘発性頭蓋内動脈瘤を調節する
要約
頭蓋内動脈瘤(IA)におけるいくつかの長鎖ノンコーディングRNA(lncRNA)の役割は、多くの研究で調査されています。この研究の目的は、血管内皮細胞傷害誘発性IAにおけるlncRNA転移関連肺腺癌転写物1(MALAT1)/ microRNA-143(miR-143)/血管内皮増殖因子-A(VEGFA)シグナル軸のメカニズムを解明することです。 。 IA組織および正常な動脈組織におけるMALAT1、miR-143、およびVEGFAの発現が検出されました。組織中のマトリックスメタロプロテイナーゼ9(MMP-9)、血清および組織中のフォンウィルブランド因子(vWF)、および血清中のエンドセリン-1(ET-1)が検出されました。モデル化されたIAラットに、血管内皮損傷を検出するために、沈黙または過剰発現したMALAT1を注射しました。 IA患者の血管内皮細胞を抽出し、サイレンシングまたは過剰発現したMALAT1をトランスフェクトして、細胞の生存率とアポトーシスに対するMALAT1の影響を検証しました。 MALAT1、miR-143、およびVEGFA間の接続は、オンライン予測、ルシフェラーゼ活性、およびRNAプルダウンアッセイによって検証されました。 MALAT1とVEGFAの過剰発現、およびmiR-143の発現不良がIA組織で見られました。 MALAT1のダウンレギュレーションは、血圧、ET-1、vWF、およびMMP-9の発現、ならびにIAのラットの血管内皮細胞のアポトーシス指数を阻害しました。ダウンレギュレーションされたMALAT1は、アポトーシスを抑制し、IAの血管内皮細胞の生存率を促進しました。 MALAT1はmiR-143およびmiR-143に結合してVEGFAを標的としました。この研究は、MALAT1がmiR-143への競合的結合を介してVEGFAの発現を高め、それによってアポトーシスを促進し、IAの血管内皮細胞の生存率を低下させることを示唆しています。
はじめに
頭蓋内動脈瘤(IA)は、脳動脈瘤としても知られ、脳動脈腔または動脈壁の局所的な異常な拡大によって誘発される頭蓋内圧の上昇によって引き起こされます[1]。 IAは高い死亡率と罹患率を伴う重篤な疾患であり、有病率は一般人口で約1〜3%です[2]。 IAの主な臨床的特徴は、脳血管れん縮、自発性脳出血、および動眼神経麻痺です[3]。これまでのところ、IAの発生と発症につながる一般的な危険因子には、血行力学的障害、遺伝子、老化、感染症、および先天性因子が含まれます[4]。主な臨床治療、主にクリッピング術および/または血管内コイル塞栓術は、動脈瘤の破裂を防ぐために機能します[5]。ただし、IAの管理におけるより効果的な方法の緊急の必要性を反映して、IAの根底にある詳細なメカニズムはまだ解明されていません。
長鎖ノンコーディングRNA(lncRNA)は、200ヌクレオチドより長く、ノンコーディングRNAの一種に属します[6]。 IAにおけるlncRNA増殖停止関連lncRNA1の発現はダウンレギュレートされていると報告されています。腺癌転写物1(MALAT1)は、長さが約8000ntの高度に濃縮され広く発現しているlncRNAです[7]。 MALAT1は、胸部大動脈瘤の平滑筋機能障害を調節することが報告されています[8]。また、ある研究では、IAにおけるlncRNAとメッセンジャーRNA(mRNA)の異常な発現が示され、lncRNA-mRNA共発現ネットワークは、IAの病因を見つける手がかりを提供します[9]。 MALAT1は、ヒト骨髄間葉系幹細胞におけるmiRNA-143(miR-143)の発現を標的とすることにより、オステリックスの発現を促進し、骨形成分化を調節することが示唆されています[10]。ある研究では、平滑筋細胞におけるクルッペル様因子5の調節と、IAにおけるその収縮性および増殖の逆転におけるmiR-143 / 145クラスターの役割も示されています[11]。 Feng et al。によると、循環中のmiR-143 / 145およびより高いマトリックスメタロプロテイナーゼ-9(MMP-9)レベルのダウンレギュレーションは、IAの形成および破裂に関連している可能性があります[12]。分析の結果、最も制御されていないmiRNA(miR-143およびmiR-145)は、血管内皮増殖因子(VEGF)などのシグナル経路である標的遺伝子や細胞の動きや接着を調節する他の遺伝子に共通していることが明らかになりました[13]。 。ある研究により、IAにおける血管内皮成長因子-A(VEGFA)の変動の予測的重要性が明らかになりました[14]。したがって、血管内皮損傷によって誘発されるIAにおけるMALAT1 / miR-143 / VEGFAシグナル軸のメカニズムを評価しようとしました。
材料と方法
倫理声明
この研究は、中国科学技術大学生命科学医学部のUSTC第一付属病院の倫理委員会によって承認され、ヘルシンキ宣言の信条に従った。参加者は、この研究において書面によるインフォームドコンセントを提供した。すべての動物実験は、国立衛生研究所の実験動物の管理と使用に関するガイドに沿ったものでした。この議定書は、中国科学技術大学生命科学医学部のUSTC第一付属病院の動物実験倫理委員会によって許可されました。
調査対象
私たちの実験には、画像検査によって診断され、中国科学技術大学生命科学医学部のUSTC第一付属病院で脳神経外科クリッピングを受けた20人のIA患者(IAグループ)が選ばれました。 43.27±6.25歳の男性11人と女性9人がいました。 IA組織がサンプリングされました。一方、側頭極からの側頭皮質動脈血管組織は、扁桃体と海馬硬化によって引き起こされた側頭葉てんかんの20人の患者(対照群)から切除されました。切除された組織は、手術後の組織病理学的検査により正常な動脈組織として検査され、44.18±5.91歳の男性13人と女性7人がいた。 IA群と対照群( P の両方)の間で、性別と年齢に有意差は認められませんでした。> 0.05)。静脈血サンプル(2本のチューブ)は、手術前の朝に同時に絶食状態のすべての被験者から採取されました。
IAのラットモデルの確立
体重200〜250 gの60匹のクリーングレードのSprague-Dawley雄ラット(Hunan SJA Laboratory Animal Co.、Ltd.、Hunan、China)を7日間飼育しました(25±2°C、相対湿度65〜70)。 %、12時間の明暗サイクル、自由な水と食物摂取)。ラットをブタ膵臓エラスターゼとともに外頸動脈および分岐動脈壁の周りに滴下した。外頸動脈は、外頸動脈の枝で約1.5mmの2本の手術ラインで結紮されました。外頸動脈を2本の線の間で切断して、外頸動脈のブラインドセグメントに内頸動脈瘤を形成した。手術後1週間、ラットに1%生理食塩水を与えた。 1ヶ月後に大脳血管造影を行い、動脈瘤の形成を観察した。
IAラットモデルの確立後、50匹のラットをランダムにブランクグループ( n )に分配しました。 =10、モデル化されたラットは、100μLのリン酸緩衝生理食塩水(PBS)を1日1回定位注射し、ショートヘアピンRNA(sh)陰性対照(NC)グループ( n =10、モデル化されたラットは、100μLのsh-MALAT1 NCを1日1回定位注射で治療されました)、sh-MALAT1グループ( n =10、モデル化されたラットは、100μLのsh-MALAT1プラスミドの定位注射で1日1回処理されました)、過剰発現(Oe)-NCグループ( n =10、モデル化されたラットは、100μLのOe-MALAT1 NCプラスミドを1日1回定位注射し、Oe-MALAT1グループ( n =10、モデル化されたラットは、100μLのOe-MALAT1プラスミドの定位注射で1日1回治療されました)[15]。上記のプラスミドは、Shanghai Genechem Co.、Ltd。(Shanghai、China)によって作成されました。
ラットの血圧テスト
ラットの尾動脈の血圧は、手術後1、4、12週目に測定されました。血圧を測定する前に、外温の乱れを防ぐために、ラットを一定温度の加熱装置にしばらく置いた。第二に、活動の干渉を防ぐために、ラットを特別なラットケージで数分間静かに保った。血圧が大きく変動する場合は、異なる時間に2〜3回測定して、平均値を求めました。
動脈瘤組織の取得
3ヶ月後、ラットを腹腔内注射により1%ペントバルビタールナトリウム(40 mg / kg)で麻酔し、静脈から血液サンプルを採取しました。ラットを安楽死させ、胸を開き、左心室を大動脈に挿管し、そして空洞を切断して血液を放出した。一方、ヘパリンナトリウムを含む30 mLの生理食塩水を急速な心臓灌流に利用して血液を洗い流した後、10 mLの10%ポリホルムアルデヒド/0.1 M PBS(pH 7.4)を脳に注入しました。灌流および固定後、ラットの脳を開いた。頭蓋底の動脈循環を注意深く観察し、動脈瘤組織を分離し、動脈瘤の変化を顕微鏡で観察しました。
酵素結合免疫吸着測定法(ELISA)
血清関連指数はELISAキットによってテストされました。採取した血液サンプルを37℃のサーモスタットに1時間入れ、3000 r / minで10分間遠心分離しました。エンドセリン-1(ET-1)およびフォンウィルブランド因子(vWF)の発現の検出は、キットの指示に従って行われました(すべてのキットは、中国江蘇省の南京江蘇生物工学研究所から購入しました)。
ヘマトキシリン-エオシン(HE)染色
サンプルを10%ホルマリンで24時間以上固定し、パラフィンブロックで保存しました。パラフィンブロックをキシレンで20分間脱ロウし、アルコールの勾配下降系列(100%、95%、80%、75%)で1分間脱水し、ヘマトキシリンで10分間染色しました。次に、組織を蒸留水ですすぎ、塩酸エタノールで30秒間分化させ、50°Cの温水に5分間浸しました。エオシン溶液で染色し、組織を蒸留水ですすぎ、70%および90%のアルコールで脱水し、キシレンで透明にし、中性ガムで密封しました。組織の形態を高倍率の顕微鏡で観察しました。
透過型電子顕微鏡観察
予備の組織を2.5%グルタルジアルデヒドと1%オスミチン酸で固定し、脱水してからEpon812樹脂で包埋しました。半薄切片をトルイジンブルーで染色し、トリミングし、超薄切片にした。切片を酢酸ウラニルとクエン酸鉛で染色し、JME-2000EX透過型電子顕微鏡(日立ハイテクノロジーズ株式会社、上海、中国)で観察しました。
末端デオキシヌクレオチドトランスフェラーゼを介したデオキシウリジン三リン酸-ビオチンニックエンドラベリング(TUNEL)染色
TUNELキット(Roche、バーゼル、スイス)に基づいて、IAラットの内皮細胞アポトーシスを観察するためにTUNEL染色が暗示されました。調製したラット動脈瘤切片をキシレンで2回洗浄し(5分/時間)、アルコールの下降系列(100%、95%、80%、75%)で3回(5分/時間)脱水しました。組織をDNaseを含まない20μg/ mLプロテアーゼK溶液で15〜30分間処理し、50μLのTUNEL反応溶液で60分間滴下し、50μLのジアミノベンジジン(DAB)で25°Cで10分間現像しました。次に、切片をヘマトキシリンで対比染色し、勾配アルコールで脱水し、キシレンで透明にし、中性ガムで密封した。切片を光学顕微鏡で観察し、アポトーシス指数を計算しました。
動脈瘤血管内皮細胞の分離と同定
内皮細胞の単離は、ボスコロらによって実施された方法に従って実施された。 [16]。 IA組織を3mm 2 に切断しました。 フラグメント。組織を0.1%コラゲナーゼB / 0.1%ジスパーゼ(Roche)とともに37°Cで25分間インキュベートしました。次に、事前に剥離した組織を2 mLのピペットで2分間粉砕し、100μmのストレーナー(Thermo Fisher Scientific、ロックフォード、イリノイ州、米国)でろ過しました。続いて、細胞懸濁液を遠心分離し、次にMV2培地(成長因子および20%ウシ胎児血清を含む)(PromoCell、ハイデルベルク、ドイツ)に再懸濁した。細胞を1×10 4 で播種しました 1μg/ cm 2 でコーティングされた培養フラスコ内の細胞/ mL フィブロネクチン。ジャクソンらによって記述された方法に従う。 [17]、80〜100%のコンフルエンスの細胞を、Ulex europaeus Agglutinin I(UEA)(Vector Laboratories、Ltd.、Peterborough、UK)でコーティングされたビーズ(Dynabeads M-450 Tosylactived、Oxoid、Hampshire、UK)で分離しました。 。レクチンでコーティングされたビーズに付着した内皮細胞を磁性粒子濃縮器で集め、非結合細胞を基礎培地で洗浄した。 UEA陽性細胞を培地に再懸濁し、フィブロネクチンでコーティングした培養フラスコに播種して、細胞の接着と増殖速度を改善しました。
細胞は、フィブロネクチンでコーティングされたチャンバースライド上のMV2で増殖させた。細胞のコンフルエンスが80〜100%に達したら、細胞を4°Cのアセトンで固定し、1%Triton X-100で5分間、次に0.5%ウシ血清アルブミン(BSA)で15分間処理しました。細胞にvWFに対する一次抗体(1:300、Abcam、ケンブリッジ、マサチューセッツ州、米国)を滴下し、2時間インキュベートし(NCは一次抗体の非存在下で実施)、西洋ワサビペルオキシダーゼを推測した免疫グロブリンG(1: 150、Abcam)および30分間インキュベートしました。次に、細胞を50μLのDABで25°Cで5分間増殖させ、ヘマトキシリンで対比染色し、0.1%塩酸で分化させ、アルコールで脱水した後、キシレンクリアランスを行い、中性ガムを密封しました。乾燥後、細胞を倒立顕微鏡で撮影しました。
セルのグループ化とトランスフェクション
対数期の動脈瘤血管内皮細胞は、ブランクグループ(無治療の動脈瘤血管内皮細胞)、sh-NCグループ(sh-MALAT1NCプラスミドでトランスフェクトされた動脈瘤血管内皮細胞)、sh-MALAT1グループ(sh-MALAT1グループ)の5つのグループに割り当てられました。 sh-MALAT1プラスミドでトランスフェクトされた動脈瘤血管内皮細胞)、Oe-NCグループ(Oe-MALAT1NCプラスミドでトランスフェクトされた動脈瘤血管内皮細胞)、およびOe-MALAT1グループ(Oe-MALAT1プラスミドでトランスフェクトされた動脈瘤血管内皮細胞)。上記のプラスミドはGenechemによって合成されました。細胞トランスフェクションは、lipofectamine TM の指示に従って実施しました。 2000試薬(11668-027、Invitrogen、カールスバッド、カリフォルニア、米国)。
3-(4,5-ジメチルチアゾール-2-イル)-2,5-ジフェニルテトラゾリウムブロミド(MTT)アッセイ
各グループの血管内皮細胞は、96ウェルプレートに3×10 4 の密度で播種されました。 細胞/ mLおよび37°C、5%CO 2 で培養 48時間。各グループに5つの並列ウェルを設定し、各ウェルに20μLの新鮮なMTT溶液(5 mg / mL、Sigma、米国ミズーリ州セントルイス)を追加しました。 4時間の反応後、細胞を200μLのジメチルスルホキシドと混合しました。完全に溶解した後、各グループの細胞の光学密度値をマイクロプレートリーダー(BioRad、Hercules、California、USA)で490nmで測定しました。
フローサイトメトリー
細胞周期分布は、ヨウ化プロピジウム(PI)染色によってテストされました。血管内皮細胞を分離し、遠心分離し、予冷した75%エタノールで再懸濁し、-20°Cで一晩固定しました。細胞を遠心分離して上清を捨てた。細胞を450μLのPBSに添加し、100μLのRnase Aを添加し、光を避けて4°Cで30分間400μLのPIで染色しました。フローサイトメーター(FACSCalibur、Becton、Dickinson and Company、NJ、USA)を使用して、細胞周期の分布をテストしました。
細胞アポトーシスは、アネキシンV / PI二重染色によってテストされました。分離した内皮細胞を集め、PBSで3回洗浄した。細胞を100μLの予冷した1×結合バッファーで再懸濁し、それぞれ5μLのアネキシンおよび5μLのPIと混合しました。細胞アポトーシスはフローサイトメーターによってテストされました。アネキシンVを横軸、PIを縦軸として、左上の象限は機械的損傷細胞を表しています(AnnexinV - / PI + )、後期アポトーシス細胞または壊死細胞の右上象限(AnnexinV + / PI + )、陰性正常細胞の左下象限(AnnexinV - / PI − )、および初期アポトーシス細胞の右下象限(AnnexinV + / PI − )。総アポトーシス細胞(AnnexinV + / PI − およびAnnexinV + / PI + )が計算され、パーセンテージで表されました。
逆転写定量的ポリメラーゼ連鎖反応(RT-qPCR)
組織および細胞内の全RNAは、Trizol(Takara Biotechnology Ltd.、Dalian、China)によって抽出され、RNAの濃度と純度が決定されました。 RNAの相補的DNAへの逆転写のプロセスは、逆転写キット(K1621、Fermentas、メリーランド、ニューヨーク、米国)の指示に従って実施された。 MALAT1、miR-143、およびVEGFAプライマー配列(表1)は、Genechemによって考案および作成されました。 lncRNA、miRNA、またはmRNAの発現を評価するために、SYBR GreenPCR Master Mix(Takara、Tokyo、Japan)とRoche Real-Time PCR system(Roche)を使用してRT-qPCRを実施しました。 U6はmiR-143の内部パラメーターとして設定され、MALAT1とVEGFAは、内部パラメーターとしてグリセルアルデヒド-3-リン酸デヒドロゲナーゼ(GAPDH)を使用しました。標的遺伝子の相対的な転写レベルは、2 -△△Ct によって計算されました。 メソッド。
<図>ウエスタンブロット分析
組織および細胞からの総タンパク質が抽出されました。タンパク質濃度は、ビシンコニン酸キット(Boster Biological Technology Co. Ltd.、Wuhan、Hubei、China)の指示に従って決定されました。タンパク質は、10%ポリアクリルアミドゲル(Boster Biological Technology)を用いた電気泳動によって分離されました。メンブレンをポリフッ化ビニリデンメンブレンに転写し、5%BSAで1時間シールしました。膜を、Ki-67(1:1000)、VEGFA(1:1000)、vWF(1:1000)、およびマトリックスメタロプロテアーゼ(MMP)-9(1:1000、Abcam、ケンブリッジ、英国)に対する一次抗体とともにインキュベートしました。 、Cyclin D1(1:1000)、Bax(1:1000)、およびBcl-2(1:1000、Santa Cruz Biotechnology、Santa Cruz、カリフォルニア、米国)、およびGAPDH(1:2000、Jackson Immuno Research、Grove、ペンシルベニア、米国)およびホースラディッシュペルオキシドで標識された二次抗体(1:500、Jackson Immuno Research)。メンブレンはオデッセイの2色赤外線蛍光走査イメージングシステムで取得し、バンドのグレー値はQuantityOne画像解析ソフトウェアで測定しました。
デュアルルシフェラーゼレポーター遺伝子アッセイ
MALAT1とmiR-143の結合部位は、バイオインフォマティクスのWebサイト(https://cm.jefferson.edu/rna22/Precomputed/)によって予測され、説明されました。 MALAT1とmiR-143の結合関係は、ルシフェラーゼレポーター遺伝子アッセイによってさらに検証されました。合成MALAT13 '非翻訳領域(3'UTR)遺伝子フラグメントをpmiR-Reportルシフェラーゼレポーターベクター(Thermo Fisher Scientific)に導入し、エンドヌクレアーゼ部位Bamh1およびEcor1によってMALAT1野生型(MALAT1-WT)を生成しました。配列の相補配列変異部位をMALAT1-WTで考案し、ターゲットフラグメントをpmiR-Reportルシフェラーゼレポーターベクターに挿入して、制限エンドヌクレアーゼ消化後にT4 DNAリガーゼによってMALAT1変異型(MALAT1-MUT)を生成しました。正しくシーケンスされたMALAT1-WTおよびMALAT1-MUTは、模倣NCおよびmiR-143模倣で血管内皮細胞にコトランスフェクトされました。トランスフェクションの48時間後に細胞を回収して溶解し、ルシフェラーゼ活性をルシフェラーゼ検出キット(BioVision、サンフランシスコ、カリフォルニア州、米国)とルミノメーター(Glomax20 / 20、プロメガ、マディソン、ウィスコンシン州、米国)でテストしました。
>miR-143とVEGFAの標的関係、およびmiR-143とVEGFA 3'UTRの結合部位は、バイオインフォマティクスのWebサイト(http://www.targetscan.org/vert_72/)を通じて予測されました。 miR-143結合部位を含むVEGFA3'UTRプロモーター領域の配列を合成し、pmiR-Reportルシフェラーゼレポーターベクターにクローニングして、VEGFA-WTを生成しました。このレポーターに基づいて、VEGFA-WTとmiR-143の結合部位が変異してVEGFA-MUTを形成しました。 VEGFA-WTまたはVEGFA-MUTレポーターを模倣NCまたはmiR-143模倣と混合し、血管内皮細胞に48時間コトランスフェクトしました。その後、細胞を溶解し、ルシフェラーゼ検出キットでルシフェラーゼ活性をテストしました。
RNA-プルダウンアッセイ
miR-143とMALAT1の結合関係を検証するために、RNAプルダウンアッセイを実施しました。 3つのビオチン標識miRNA配列Bio-miR-143-WT、Bio-miR-143-MUT、およびBio-miR-NCが設計され、GenePharma Company(上海、中国)に委託されました。これらのビオチン化オリゴヌクレオチドを48時間細胞にトランスフェクトしました。次に、細胞を回収し、特定の細胞溶解物(Ambion、オースティン、テキサス、米国)と10分間インキュベートしました。その後、溶解物をRNaseフリーおよび酵母tRNA(すべてSigma製)でプレコートしたM-280ストレプトアビジンビーズで4°Cで3時間孵化し、冷溶解液で2回、低濃度で3回洗浄しました。塩緩衝液、および高塩緩衝液で1回。拮抗的なmiR-143プローブがNCとして確立されました。全RNAをTrizolで抽出した後、RT-qPCRを使用してMALAT1濃縮レベルをテストしました。
統計分析
すべてのデータは、SPSS 21.0ソフトウェア(IBM Corp. Armonk、NY、USA)によって説明されました。測定データは平均±標準偏差で表されました。 2つのグループ間の比較は、独立したサンプル t によって行われました。 複数のグループ間の比較は一元配置分散分析(ANOVA)によって評価され、ペアワイズ比較はテューキーの多重比較検定によって実装されました。 MALAT1の発現とIAの臨床病理学的特徴との関係は、カイ二乗検定によって決定されました。 P 0.05未満の値は、統計的に有意な差を示していました。
結果
MALAT1とVEGFAは過剰発現しており、miR-143はIA組織でダウンレギュレーションされています
IA群と対照群の血清中のET-1とvWFの発現はELISAによって検出され、結果は、ET-1とvWFの発現が対照群と比較してIA群で増加したことを示しました(両方 P <0.05)(図1a、b)。
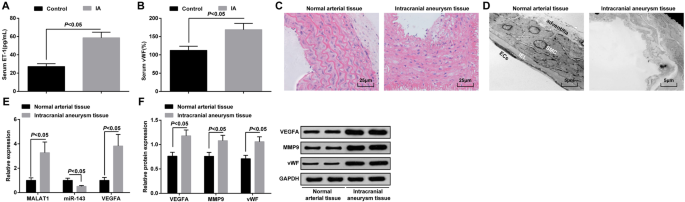
MALAT1とVEGFAは過剰発現しており、miR-143はIA組織でダウンレギュレーションされています。 a ELISAによるIA患者および側頭葉てんかん患者の血清におけるET-1発現。 b ELISAによるIA患者および側頭葉てんかん患者の血清におけるvWF発現。 c HE染色によるIA組織および正常動脈組織の病理学的観察。 d 透過型電子顕微鏡によるIA組織および正常な動脈組織の形態学的観察。 e RT-qPCRによるIA組織および正常動脈組織におけるMALAT1、miR-143、およびVEGFAmRNAの発現。 f ウエスタンブロット分析によるIA組織および正常動脈組織におけるVEGFA、MMP-9、およびvWFタンパク質の発現。内皮細胞(EC)、内部弾性板(IEL)、平滑筋細胞(SMC)。測定データは平均±標準偏差として表されました。グループ間の比較は、独立したサンプル t によって行われました。 テスト
IA組織の病理学的変化は、HE染色を通して観察された。正常な動脈組織では、内胚葉、内皮細胞、平滑筋細胞に明らかな損傷は見られず、細胞はきれいに配置され、完全な構造を持っていました。 IA組織は、損傷した内皮細胞、変性した平滑筋細胞、弱毒化した動脈壁、破裂した弾性線維、および浸潤した炎症細胞を示しました(図1c)。
IAおよび正常な動脈組織の超微細構造の変化を透過型電子顕微鏡で観察した。内皮細胞は完全であり、外膜構造は無傷であることがわかった。正常な動脈組織では、壊れた内部弾性膜やアポトーシス平滑筋細胞は見られませんでした。 IA組織では、血管壁の重度の変性が見られ、主にほとんどの内皮細胞の消失、内部弾性層の重度の破壊、平滑筋細胞の重度の損傷と劣化、血管外膜の障害として現れました。 (図1d)。
RT-qPCRは、MALAT1、miR-143、およびVEGFA mRNAの発現を測定するために実施され、一方、IA組織および正常な動脈組織におけるVEGFA、MMP-9、およびvWFタンパク質の発現のウエスタンブロット分析が行われました。正常な動脈組織とは対照的に、MALAT1、VEGFA、MMP-9、およびvWFの発現レベルが上昇し、IA組織(すべての P )でmiR-143の発現が低下することが実証されました。 <0.05)(図1e、f)。
ハントヘスグレード、内皮損傷の程度、および喫煙歴は、IA組織におけるMALAT1発現と相関しています
MALAT1の発現中央値に照らして、患者は2つのグループに割り当てられました:低発現グループと高発現グループ。 MALAT1の発現とIA患者の臨床病理学的特徴との関係を分析した。結果は、ハントヘスのグレード、内皮損傷の程度、および喫煙歴がMALAT1の発現と関連していることを示唆しました(すべて P <0.05)、年齢、性別、および手術モードはMALAT1の発現とは関連していませんでした(すべて P > 0.05)(表2)。
<図>ダウンレギュレーションされたMALAT1は、血圧、ET-1、vWF、MMP-9の発現、およびラットの血管内皮細胞のアポトーシス指数を抑制しますIA付き
表3に示すように、MALAT1のノックダウンは低下しましたが、MALAT1の回復は4週目と12週目に血圧を上昇させました。
<図>ELISAは、MALAT1のダウンレギュレーションが減少する一方で、MALAT1のアップレギュレーションがIAのラットの血清におけるET-1およびvWFの発現を上昇させることを明らかにしました(図2a、b)。
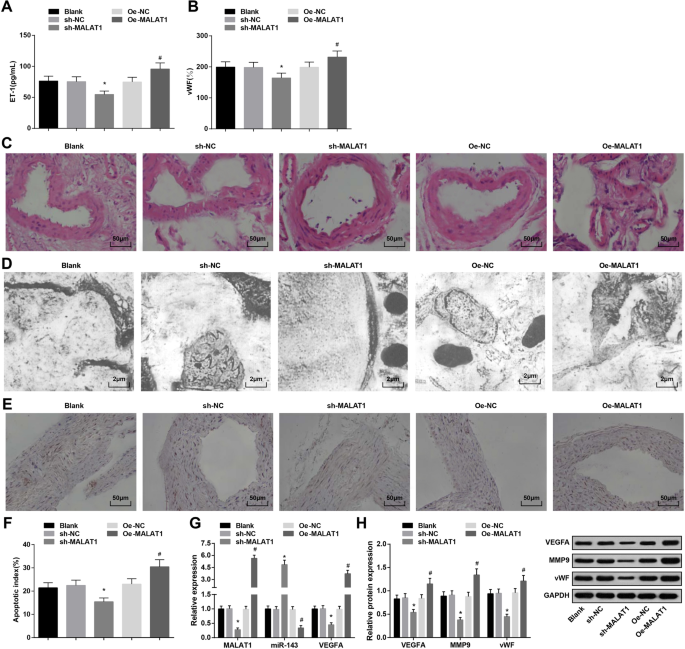
ダウンレギュレーションされたMALAT1は、血圧、ET-1、vWF、MMP-9の発現、およびIAラットの血管内皮細胞のアポトーシス指数を抑制します。 a ELISAによるラットの血清中のET-1発現。 b ELISAによるラットの血清中のvWF発現。 c HE染色で観察されたラットのIA組織の病理学的変化。 d 透過型電子顕微鏡で観察されたラットのIA組織の超微細構造。 e TUNEL染色による血管内皮細胞のアポトーシス。 f ラットの血管内皮細胞アポトーシス指数。 g RT-qPCRによるラットのIA組織におけるMALAT1、miR-143、およびVEGFAmRNAの発現。 h ウエスタンブロット分析によるラットのIA組織におけるVEGFA、MMP-9、およびvWFタンパク質の発現。 * P <0.05 vs. sh-NCグループ、# P <0.05対Oe-NCグループ。測定データは平均±標準偏差として表され、複数のグループ間の比較は、一元配置分散分析とそれに続くテューキーの多重比較検定によって評価されました
各グループのIA組織の病理学的変化をHE染色で調査した。ブランクグループ、sh-NCグループ、およびOe-NCグループは、損傷した内膜、剥離した内皮細胞、変性した平滑筋細胞、細胞と層の減少、および動脈壁の薄化を示しました。 sh-MALAT1群では、内皮細胞、内皮細胞、平滑筋細胞層、頭蓋内動脈外膜層がわずかに損傷していましたが、きれいに配置されていました。 Oe-MALAT1グループは、内膜層の消失、内皮細胞の剥離、弾性線維の破壊、炎症細胞の浸潤を示しました(図2c)。
各群のラットのIA組織を透過型電子顕微鏡で観察した。ブランク群、sh-NC群、Oe-NC群では、内皮細胞の変性、内皮下層の崩壊、内弾性層の消失、平滑筋細胞の減少が見られた。 sh-MALAT1グループは、平坦な内皮細胞、卵形の核、およびコラーゲン線維の増加を示しましたが、弾性層はありませんでした。 Oe-MALAT1グループは、内皮細胞が消失し、弾性層が筋層から分離し、内腔に崩壊したことを特徴としています(図2d)。
IAラットにおける血管内皮細胞のアポトーシス指数は、TUNEL染色によって試験された。 MALAT1をサイレンシングすると、血管内皮細胞のアポトーシス指数が低下しましたが、過剰発現したMALAT1は反対の効果を示しました(図2e、f)。
MALAT1、miR-143、およびVEGFA mRNA発現のRT-qPCR検出、およびIA組織におけるVEGFA、MMP-9、およびvWFタンパク質発現のウエスタンブロット分析は、MALAT1除去がMALAT1、VEGFA、MMP-9、およびvWF発現を抑制したことを示しました、およびmiR-143の発現を高めました。それどころか、MALAT1の上昇は、これらの遺伝子発現に反対の影響を及ぼしました(図2g、h)。
MALAT1の低発現は、IAにおける血管内皮細胞の生存率を高め、アポトーシスを抑制します
免疫組織化学的染色を使用して、内皮特異的マーカーvWFの発現を検出した。 IA血管内皮細胞の細胞質は、陽性である微細な茶色の粒子で覆われているが、そのNCグループの細胞質は、茶色の粒子を有さなかったことが明らかになった。その結果、培養細胞が内皮細胞であることが確認されました(図3a、b)。
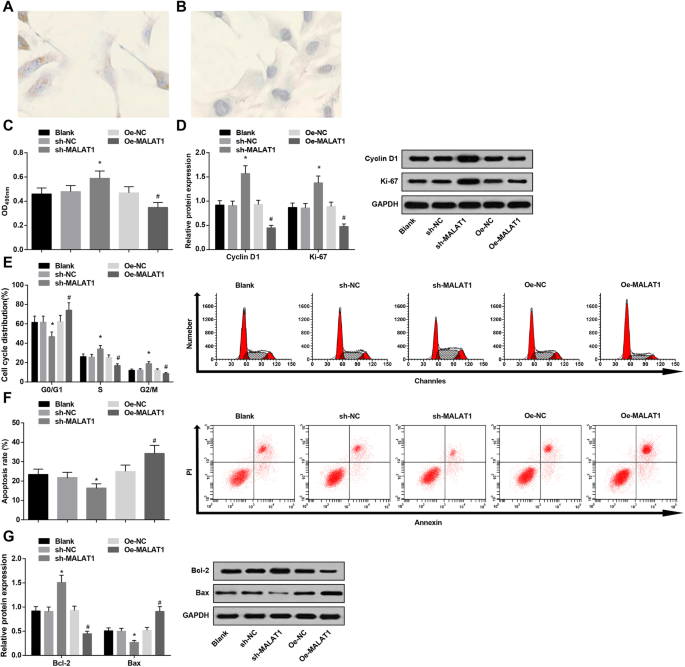
MALAT1の低発現は、IAの血管内皮細胞の生存率を高め、アポトーシスを抑制します。 a IA血管内皮細胞におけるvWF免疫組織化学的染色:IA血管内皮細胞は黄色の微粒子で覆われていた。 b IA血管内皮細胞におけるvWF免疫組織化学的染色:IA血管内皮細胞はNCグループに茶色の粒子を示さなかった。 c MTTアッセイによる各グループの血管内皮細胞の生存率。 d ウエスタンブロット分析による各グループのCyclinD1およびKi-67のタンパク質発現。 e PI染色により各群の細胞周期が変化します。 f アネキシンV / PI二重染色による各グループの細胞アポトーシス率。 g ウエスタンブロット分析による各グループのBaxおよびBcl-2タンパク質の発現。 * P <0.05 vs. sh-NCグループ、# P <0.05対Oe-NCグループ。測定データは平均±標準偏差として表され、複数のグループ間の比較は、一元配置分散分析とそれに続くテューキーの多重比較検定によって評価されました
MTTアッセイ、フローサイトメトリー、およびウエスタンブロット分析を利用して、血管内皮細胞の生存率とアポトーシスをテストしました。 MALAT1の減少は、血管内皮細胞の生存率を促進し(サイクリンD1およびKi-67の発現を高め)、アポトーシスを抑制しました(G0 / G1期の細胞を減らし、S期およびG2 / M期の細胞を増やし、Baxを減らし、Bcl-2の発現を高めました)。 。ただし、MALAT1のアップレギュレーションは、細胞の生存率とアポトーシスに対するMALAT1の減少とは逆の方法で機能しました(図3c–g)。
MiR-143はMALAT1にバインドされており、VEGFAはmiR-143のターゲット遺伝子です
各グループの血管内皮細胞におけるMALA1、miR-143、およびVEGFAの発現は、RT-qPCRおよびウエスタンブロット分析によって検証されました。結果は、MALA1ノックダウンがMALA1とVEGFAの発現を抑制し、miR-143の発現を増強することを示しました。それどころか、MALA1のアップレギュレーションは、MALAT1とVEGFAの発現を増加させ、miR-143の発現を減少させました(図4a、b)。
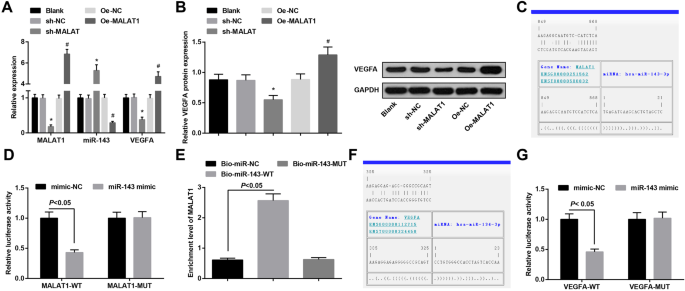
MiR-143はMALAT1に結合しており、VEGFAはmiR-143の標的遺伝子です。 a 各グループの動脈瘤の血管内皮細胞におけるMALAT1、miR-143、およびVEGFAmRNAの発現。 b 各グループの動脈瘤の血管内皮細胞におけるVEGFAタンパク質の発現。 c バイオインフォマティクスのウェブサイトによって予測されたMALAT1とmiR-143の結合部位。 d デュアルルシフェラーゼレポーター遺伝子アッセイによって検証されたMALLA1とmiR-143の調節関係。 e RNAプルダウンアッセイによって検証されたMALAT1とmiR-143の間の結合関係。 f バイオインフォマティクスのウェブサイトによって予測されたmiR-143とVEGFAの結合部位。 g デュアルルシフェラーゼレポーター遺伝子アッセイによって検証されたmiR-143とVEGFAの調節関係。 * P <0.05 vs. sh-NCグループ、# P <0.05対Oe-NCグループ。測定データは平均±標準偏差として表され、2つのグループ間の比較は独立したサンプル t によって評価されました。 テスト、および複数のグループ間の比較は、一元配置分散分析とそれに続くテューキーの多重比較検定によって評価されました
MALAT1とmiR-143の間の特異的結合領域は、オンライン分析ソフトウェアによって決定されました(図4c)。デュアルルシフェラーゼレポーター遺伝子アッセイの結果は、ルシフェラーゼ活性がMALAT1-WT + miR-143模倣グループとMALAT1-WT + mimic-NCグループで損なわれていることを明らかにしました( P <0.05)。ただし、MALAT1-MUT + miR-143ミミックグループとMALAT1-MUT + mimic-NCグループのルシフェラーゼ活性に明確な違いはありませんでした( P > 0.05)、miR-143がMALAT1に特異的に結合したことを示します(図4d)。 RNAプルダウンアッセイの結果は、Bio-miR-143-WTグループのMALAT1の濃縮レベルがBio-miR-NCグループと比較して高くなったことを報告しました( P <0.05)ですが、Bio-miR-143-MUTグループ( P )のMALAT1濃縮レベルに明確な違いはありませんでした。> 0.05)(図4e)。
バイオインフォマティクスソフトウェアは、miR-143とVEGFAの間のターゲットとなる関係を分割しました(図4f)。ルシフェラーゼ活性の結果は、VEGFA-WTとmiR-143が血管内皮細胞にコトランスフェクトされた後に相対的なルシフェラーゼ活性が抑制されることを示しました( P <0.05)。ただし、血管内皮細胞の相対的なルシフェラーゼ活性は、VEGFA-MUTとmiR-143-mimic( P )の同時トランスフェクションの影響を受けませんでした。> 0.05)(図4g)。 VEGFAはmiR-143の直接の標的遺伝子であることが示された。
ディスカッション
IAは深刻な頭蓋内疾患であり、主にくも膜下出血を引き起こします[18]。以前の研究では、IAにおけるlncRNA関連の競合的内因性RNAネットワークの関与が実証されています[19]。また、最近の研究は、miR-143 / 145遺伝子プロモーター領域の機能的多型がIAのリスクと関連しているという証拠を提供しました[20]。 Xuらが実施した研究では。 、VEGFAなどの血管新生因子の過剰発現がIAの形成と破裂に関連している可能性があることが示されています[21]。 IAにおけるMALAT1の分子メカニズムを説明するために、一連のアッセイが実施され、その結果、血管内皮損傷によって誘発されたIAがMALAT1 / miR-143 / VEGFAシグナル軸によって調節されていることが明らかになりました。
まず、私たちの研究は、MALAT1とVEGFAがアップレギュレーションされ、miR-143がIA組織でダウンレギュレーションされているという実質的な証拠を提供しました。最近の研究では、MALAT1が脳虚血の過程で最もアップレギュレーションされたlncRNAの1つであることが示されています[22]。別の研究では、MALAT1が卵巣癌細胞でアップレギュレーションされており、卵巣癌細胞のアポトーシス、遊走、増殖のプロセスに関与することを意図していることが示されています[23]。破裂していないIAと破裂したIAの発現プロファイルに関する研究では、破裂した動脈瘤ではVEGFAなどの血管新生因子の発現がアップレギュレーションされていることが示されています[21]。さらに、臨床研究では、IA患者のmiR-143 / 145クラスターが健康な被験者と比較してダウンレギュレーションされていることが示されています[11]。さらに、miR-143 / 145は動脈瘤形成に関連するさまざまな生物学的プロセスに関与し、IA患者ではダウンレギュレーションされることが以前に議論されています[20]。これらの調査結果はすべて、以前の調査結果とほぼ同じです。
上記の発見を除いて、この研究はまた、機能獲得および機能喪失アッセイを通じて、IAにおけるMALAT1の機能的役割を調査しました。 MALAT1のダウンレギュレーションは、血圧、ET-1の血清レベル、およびIA組織におけるvWFとMMP-9の発現を低下させたと要約できます。 MALAT1のダウンレギュレーションは、グルコース誘導性のET-1転写産物のアップレギュレーションを抑制することが以前に示唆されています[24]。また、異所性のMALAT1発現がvWF生成の誘導因子であることが報告されています[25]。別の研究では、骨髄由来マクロファージにおけるMALAT1の枯渇がMMP-9の発現を阻害することが確認されています[26]。
また、IAにおけるMALAT1の機能をさらに確認するために細胞実験を行った。結果は、MALAT1ノックダウンがIAの血管内皮細胞の生存率を促進し、アポトーシスを抑制したことを示唆しています。同様に、MALAT1の障害は大動脈壁の構造を改善し、動脈瘤の成長を遅らせることが報告されています[8]。私たちの研究結果を補足するものとして、MALAT1の発現不良がアポトーシスを誘発し、急性骨髄性白血病細胞の増殖を抑制することを強調する研究があります[27]。別の研究でも、ヒト変形性関節症の軟骨細胞の増殖に対するMALAT1ノックダウンの阻害効果が実証されています[28]。それに加えて、以前の研究では、MALAT1のダウンレギュレーションがアポトーシスを誘発し、神経膠腫細胞の増殖を弱める可能性があることが一般的に確認されています[29]。さらに、RNA干渉によって誘導されるMALAT1の低発現は、アポトーシスを促進し、多発性骨髄腫細胞の増殖を抑制します[30]。まとめると、これらの研究は、IAにおけるMALAT1の分子メカニズムをある程度説明しています。
さらに、この研究は、miR-143がMALAT1に結合し、VEGFAがmiR-143の標的遺伝子であることを証明しています。同様に、ある論文は、MALAT1がmiR-143に直接結合し、その発現を抑制すると主張しています[10]。朱ら。 MALAT1は子宮頸癌細胞でmiR-143と相互作用することでその役割を果たしていることを発見しました[31]。さらに、MALAT1がmiR-200b-3pを介して間接的にVEGFAを調節することが確認されています[32]。さらに、別の研究では、miR-143-3pがVEGFAを含む血液中の多くのタンパク質標的に対するZFPM2効果を媒介することが示唆されています[33]。それにもかかわらず、IAにおけるMALAT1、miR-143、およびVEGFA間の相互作用は議論されておらず、さらなる研究が必要です。
結論
これらの結果から、MALAT1ノックダウンはmiR-143 / VEGFA軸を調節することにより、アポトーシスを抑制し、IAの血管内皮細胞の生存率を促進することが明らかです。共発現ネットワークは、VEGFAの関与によるMALAT1とmiR-143の間の接続を示唆しています。この研究の結果は、IAの開始と進行の病因を部分的に明らかにしており、研究されたターゲットは、別の観点からIAの病理を明らかにするための特に潜在的なエントリポイントである可能性があります。限定的に、IAにおけるMALAT1 / miR-143 / VEANA軸のメカニズムを包括的に説明するには、さらに大規模な研究が必要です。
ナノマテリアル