


金ナノ粒子で阻害されたチトクロームP4503A4活性と、ヒト肝細胞癌細胞株C3Aにおけるその細胞毒性の根底にある分子メカニズムの評価
要約
カチオン性分岐ポリエチレンイミン(BPEI)、アニオン性リポ酸(LA)、または中性ポリエチレングリコール(PEG)で機能化された40および80 nmの金ナノ粒子(AuNP)と、ヒト肝細胞癌(HCC)細胞株C3Aとの相互作用がヒト血漿タンパク質コロナ(PC)の有無。 80 nm LA-AuNP以外のすべての裸(PCなし)のAuNPはC3Aに対して細胞毒性がありましたが、PCはそれらの細胞毒性を弱めました。 AuNPの時間依存的な細胞取り込みは40nm BPEI-AuNP以外に増加しましたが、PCは80 nmPEG-AuNP以外の取り込みを抑制しました。 BPEI-AuNPによる酸化/ニトロソ化ストレスの二相性応答はC3A細胞で発生しましたが、PEG-AuNPは強力な抗酸化剤でした。すべての裸のAuNPは、サイズと表面電荷に関係なく、シトクロムP450(CYP)3A4活性を阻害しましたが、PCはPEG-AuNP以外にその活性を回復しました。 40 nm PEG-AuNPで調節される遺伝子発現は、主にミトコンドリアの脂肪酸β酸化と、程度は低いものの肝排出/取り込みトランスポーターに関与していました。これらの研究は、AuNPと主要な生物学的プロセスとの相互作用およびHCCにおけるそれらの基礎となる分子メカニズムの理解を深めるのに役立ちます。これは、HCC治療におけるより効果的な治療標的の開発にさらに関係している可能性があります。
背景
肝細胞癌(HCC)は、世界で最も一般的な癌の1つであり、米国で最も急速に増加している癌による死亡原因です[1、2]。 HCCが進行段階で診断されていることを考えると、治癒的HCC治療には、初期腫瘍発生時の肝移植または外科的切除、および進行状態の腫瘍に対する化学療法および放射線療法が含まれます。 HCCは、全身性の副作用を引き起こす可能性のある非選択的な細胞毒性分子である従来の抗腫瘍薬に対して高い耐性を示すことがよくあります。遺伝子治療の最近の進歩、すなわちRNA干渉(RNAi)ベースの遺伝子治療は、現在のHCC治療で利用されています[3、4]。 RNAiの有効性には、ベクターを標的細胞の内部に送達する必要があります[5]。遺伝子送達を成功させるためのベクターは、ウイルスベクターと非ウイルスベクターです。ウイルスは遺伝子送達のより高い効率を提供しますが、ウイルスベクターの安全性の懸念から非ウイルスベクターが好まれます。標的遺伝子送達または薬物送達システム用の非ウイルスベクターとしてのナノ粒子(NP)は、治療効率を改善し、HCC治療における全身および/または細胞レベルでの毒性を低下させるために大きな注目を集めています[4、6]。したがって、標的細胞および組織におけるNPの細胞障害および毒性の根底にある分子メカニズムおよび生物学的経路を特定することが非常に重要になります。最近のinvitro研究は、細胞および生化学的応答と組み合わせた遺伝子発現プロファイリングが、細胞の摂動および潜在的なNP毒性の直接評価を提供したことを示しました[7,8,9,10]。
金ナノ粒子(AuNP)は、その独特の物理化学的特性と表面化学により、単独で、または他の薬物と組み合わせて、遺伝子サイレンシング部分の標的特異的送達のための送達媒体として使用されてきました[11、12]。 AuNPと血漿タンパク質との相互作用によりタンパク質コロナが形成され、これがNP表面の化学的性質を変化させ、細胞への取り込みや潜在的な毒性などのその後の生物学的反応に影響を与えます[13、14]。さまざまなヒト癌細胞株および一次電池におけるAuNPの細胞取り込みは、サイズや表面電荷に関係なく、タンパク質コロナ形成によって重大な影響を受けました[7、8、9、14、15、16、17]。
サイズおよび表面電荷に依存する酸化ストレスは、ヒト乳癌細胞株、MDA-MB-231、肝細胞癌HepG2、およびNP細胞毒性に関連するAuNPに応答したヒト白血病HL-60細胞でも観察されました[18 、19]。 AuNPによって誘発される細胞毒性は、さまざまなヒト癌細胞株および初代ヒト細胞で細胞型特異的に発生しました[7、8、9、20、21]。
チトクロームP450(CYP)酵素は、多数の細胞毒性薬の生物活性化または不活性化に重要な役割を果たし、抗がん剤の発がん性に対する宿主の感受性を高めます[22]。 AuNPは、invivoおよびinvitroで細胞および分子レベルでCYP酵素の触媒活性に影響を及ぼしました[7、23、24、25]。 AuNPは、ヒト肺線維芽細胞株MRC-5の酸化ストレスマーカー、およびヒト臍帯静脈細胞(HUVEC)とヒト肝細胞のミトコンドリア機能障害に主に関与する差次的遺伝子発現をかなり示しています。高い細胞毒性[8、9、26]。この知識は、AuNPがさまざまな細胞型でアポトーシスまたは壊死性細胞死を引き起こし、ストレス応答経路および毒性における差次的遺伝子発現と組み合わされた細胞および生化学的機能を変化させることを相互に示唆していますが、AuNPが細胞内または生物学的内で毒性効果を発揮する特定の経路システムは不明のままです。
ここでは、この研究は、ヒトHCC細胞C3AとのAuNP相互作用に対するタンパク質コロナ、サイズ、および表面電荷の影響を調査しました。主に、C3A細胞におけるカチオン性BPEI、アニオン性リポ酸(LA)、または中性ポリエチレングリコール(PEG)で機能化された40および80 nm AuNPの時間依存性細胞取り込みが、ヒト血漿タンパク質コロナ(PC)の有無で測定されました。次に、AuNPが誘導する細胞毒性と活性酸素種(ROS)/活性窒素種(RNS)の生成を、CYP3A4活性に対する阻害効果とともにモニターしました。最後に、AuNP毒性に関連する分子作用機序は、Human Molecular Toxicology PathwayFinderとHumanDrug Transporters RT 2 を使用して特徴づけられました。 Profiler™PCRアレイ。
メソッド
金ナノ粒子の合成
40および80nmのカチオン性BPEI、アニオン性LA、およびニュートラルPEGBiopure™AuNPは、nanoComposix(San Diego、CA)からカスタム合成されました。粒子サイズ、多分散度指数(PDI)、およびゼータ(z)-電位およびスペクトル特性は、動的光散乱(DLS)、透過型電子顕微鏡(TEM)、およびUV-Vis分光法で特徴づけられました。 AuNPは、炭酸カリウム水溶液中でテトラクロロ金酸水素(III)水和物を還元した後、エージングプロセスとタンジェンシャルフローろ過(TFF)を行うことで合成されました。 AuNP表面は、ジヒドロリポ酸(0.2:1、 w )を添加することにより、LAまたはPEGで機能化されました。 / w )またはチオール-メトキシ末端PEG(Laysan Bio Inc.、アラバマ州アラブ)(0.5:1、 w / w )、それぞれ、TFF洗浄と滅菌ろ過が続きます。 AuNPのBPEI官能化表面は、LAのカルボン酸をBPEIの遊離アミンに結合させた後、TFF洗浄し、続いて遠心分離して非結合BPEIを除去することにより、EDC化学によって合成されました。
タンパク質コロナの準備
プールされたヒト血漿( n =5)は、Biological Specialty Corp.(ペンシルベニア州、コルマール)から入手しました。 AuNPはヒト血漿中でインキュベートされました(55%、 v / v )報告されているように、37°Cで1時間、250 rpmの一定速度で[7、8]。 20,000× g のリン酸緩衝生理食塩水(PBS)で繰り返し洗浄することにより、結合していない弱く結合したタンパク質を除去しました。 20°Cで20分間。最終的なヒト血漿タンパク質コロナ(PC)でコーティングされたAuNPをPBSに分散させ、さらに物理化学的特性評価または投与のために細胞培養培地で希釈しました。詳細なプロトコルは、追加ファイル1に記載されています。
AuNPの物理化学的特性評価
流体力学的直径( D H )、PDI、および脱イオン(DI)水中のBPEI、LA、およびPEGで機能化された40および80 nmの裸(PCなし)AuNPのz電位を、Zetasizer Nano ZS(Malvern)を使用して25°C、0時間で分析しました。 Instruments、ウスターシャー、英国); PCでコーティングされたAuNPのPBS、25°C、0時間。完全細胞培養培地中のすべての裸およびPCAuNPについて、37°C、0時間および24時間。完全細胞培養培地には、10%FBS(ATCC ® )を添加したイーグル最小必須培地(EMEM)が含まれていました。 、マナッサス、バージニア州)。サンプルは、それぞれ10秒の11回のサブランで5回測定されました。さらに、Synergy H1ハイブリッドマルチモードマイクロプレートリーダー(BioTek Instruments Inc.、バーモント州ウィヌースキ)を使用して、室温で0時間の吸光度スペクトルを測定しました。
透過型電子顕微鏡
AuNPの形態はTEMを使用して特徴づけられました。すべての裸のPCAuNP溶液(5μL)を200メッシュの銅グリッドに配置し、室温で風乾しました。サンプルは、Oxford検出器(FEI Company、オレゴン州ヒルズボロ)を備えたTecnai G2 Spirit BioTWINで、加速電圧120kVで観察されました。 GATAN顕微鏡スイート(GATAN Inc.、カリフォルニア州プレザントン)はAuNPの直径を測定しました。
細胞培養と生存率の測定
ヒト肝細胞癌C3A細胞(ATCC ® CRL-10741™)はATCC ® から購入しました (バージニア州マナッサス)、完全EMEM(ATCC ® )で培養 、バージニア州マナッサス)10%FBSを補充し、4日ごとに培地を交換してT75フラスコで約80%のコンフルエンスに拡大しました。 0.25%後( w / v )トリプシン–0.53 mMエチレンジアミン四酢酸(EDTA)消化、細胞を96ウェルプレートに8×10 4 で播種しました。 ウェルあたりの細胞数を95%、CO 2。の加湿雰囲気下、37°Cで培養 48時間のインキュベーション後、PCの非存在下および存在下で細胞にAuNPを投与しました。 9継代から12継代までのC3A細胞を投与に使用しました。
C3Aの実行可能性は、alamarBlue ® を使用して決定されました。 記載されている生存率アッセイ(Thermo Sci。、Waltham、MA)[7、27]。 96ウェルプレートの細胞を40および80nm BPEI-、LA-、およびPEG-AuNPで処理し、PCを使用した場合と使用しない場合の範囲は0〜250μg / cm 2 。 24時間後、alamarBlue ® の10% 完全なEMEMの試薬( v / v )を細胞培養に加え、37℃で3時間インキュベートしました。完全なEMEMは分散剤として機能しました。 AuNPとalamarBlue ® の有効成分との相互作用 試薬、レサズリンまたは還元生成物、レソルフィンを対照として測定した。 AuNPとレサズリン(細胞なし)または維持培地(細胞なし)がバックグラウンドコントロールとして機能しました。細胞生存率に比例する蛍光は、コントロールに対して正規化され、コントロール細胞グループに対するパーセンテージとして表されました。
誘導結合プラズマ質量分析による細胞取り込み測定
細胞を8×10 4 で播種しました 96ウェルプレートのウェルあたりの細胞数と無毒濃度1.56μg/ cm 2 0.5、1、3、6、12、および24時間のすべてのベアおよびPCAuNPの以前に報告されたように[28]、細胞膜に結合したAuNPとウェルへのその非特異的結合を除去するためにエッチングステップが組み込まれました。細胞採取物を王水で乾燥および消化し、NexION ™ を使用して細胞内Au濃度を定量化しました。 350X誘導結合プラズマ質量分析(ICP-MS)(PerkinElmer、マサチューセッツ州ウォルサム)。 AuNPの細胞取り込みは、以前に報告されたように計算され、細胞あたりのAuNPの数として表されました[29]。詳細なプロトコルは、追加ファイル1に記載されています。
酸化/ニトロソ化ストレス測定
細胞を8×10 4 で播種しました 96ウェルプレートのウェルあたりの細胞数と、最大125μg/ cmの40nmの裸のBPEI-およびPEG-AuNPを投与 2 1、3、24時間。酸素/ニトロソ化ストレスの直接測定は、前述のように[30]、総活性酸素種(ROS)/スーパーオキシド(SO)アッセイキット(Enzo Life Sciences、ニューヨーク州ファーミングデール)を使用してアッセイしました。 ROS /活性窒素種(RNS)(Ex488 / Em520 nm)またはSO(Ex550 / Em610 nm)の増加に比例する蛍光をマイクロプレートリーダーで測定しました。詳細なプロトコルは、追加ファイル1に記載されています。
シトクロムP4503A4アクティビティ
CYP3A4活性に対する40および80nmのベアおよびPCAuNPの悪影響は、完全に説明されているようにP450-Glo™アッセイ(Promega Corp.、ウィスコンシン州マディソン)を使用して特徴付けられました[7]。 96ウェルプレートのC3A細胞は、半数致死量(LC 50 )値:127.3μg/ cm 2 40 nm BPEI-AuNP、205.5μg/ cm 2 80 nm BPEI-AuNP、192.5μg/ cm 2 40 nm LA-AuNP、および129.5μg/ cm 2 40 nmPEG-AuNPのLC 50 以降 80 nm LA-およびPEG-AuNPの値は決定されておらず、細胞はLC 50 で処理されました。 40 nm LA-およびPEG-AuNPの値(192.5μg/ cm 2 および129.5μg/ cm 2 、 それぞれ)。 24時間のインキュベーションが終了した後、細胞をCYP3A4基質(ルシフェリン-IPA)とともに37°Cで3時間インキュベートしました。酵素活性に比例する発光シグナルをマイクロプレートリーダーで測定し、コントロールに対して正規化しました。コントロールは、AuNPと親基質または代謝物および無細胞基質との相互作用を評価するために割り当てられました。 CYP活性は、対照細胞群に対するパーセンテージとして表されました。
遺伝子発現プロファイリング
毒性のある40nm PEG-AuNPは、高い細胞取り込みを示すC3A細胞のCYP3A4活性と抗酸化活性の阻害に使用されたため、その毒性と異なる細胞応答の根底にある作用の分子メカニズムを特徴づけるために選択されました。細胞を2.5×10 6 で播種しました 6ウェルプレートのウェルあたりの細胞数とLC 50 37°Cで24時間の40nmPEG-AuNPの値。インキュベーションの最後に、細胞をRNA単離し、次に、平均RNA完全性数(RIN)値が7.8のトータルRNAを使用したcDNA合成を前述のように実施しました[7、8、9]。得られたcDNAをRT 2 と混合しました SYBRグリーンマスターミックス(Qiagen Inc.、カリフォルニア州バレンシア)を使用して、Human Molecular Toxicology PathwayFinderまたはHumanDrug Transporters RT 2 に適用します。 Quantstudio™7Flex(Applied BioSystem、カリフォルニア州フォスターシティー)のProfiler™PCRアレイ。倍率変化<−2および> 2および p を伴う差次的に発現する遺伝子 <0.05は、目的の遺伝子のダウンレギュレーションとアップレギュレーションを表しています。 RT 2 を検証するには PCRアレイデータ、9つの選択された遺伝子の発現は、cDNA合成とそれに続くリアルタイムPCRで評価されました。プライマー配列は、追加ファイル1:表S1にまとめられています。すべてのPCR反応は3回行った。リアルタイムPCR条件と定量化の詳細なプロトコルは、追加ファイル1に記載されています。
統計分析
半数致死量(LC 50 )C3A細胞のAuNPの値は、GraphPad Prism 6(La Jolla、CA)を使用して、観測データ(AuNP濃度レベルと対応する細胞生存率の入力)に可変勾配のヒルの式を当てはめることによって推定されました[7]。一元配置分散分析(ANOVA)は、SAS 9.4(SAS Institute、ノースカロライナ州ケアリー)を使用して実施し、C3A細胞におけるROS / RNS産生および細胞取り込みに対するAuNPの影響を評価しました。有意である場合は、 p でテューキーの正直有意差(HSD)検定を使用して多重比較を実行しました。 <0.05。
結果と考察
裸およびヒト血漿PCAuNPの物理化学的特性評価
流体力学的直径(D H )に対するAuNP周辺のNPサイズ、表面電荷、およびヒト血漿PC形成の影響 )、多分散度指数(PDI)、zポテンシャル、スペクトル特性、および形態は、DLS、TEM、およびUV-Vis分光法を使用して特徴付けられています(図1)。 TEM画像では、DI水中のすべての裸(PCなし)のAuNPは単分散であり、サイズ分布が狭く、独特のUV-Visスペクトル範囲が521〜553 nmでした(図1a)。 AuNP周辺のPC形成は、サイズ分布の変化と吸収スペクトルの赤方偏移とともに観察されました(図1b)。 D H 完全なEMEMの40および80nmベアおよびPCAuNPのPDI値は、PDI値の減少(それぞれ0.29および0.32)を示した40および80 nm PC BPEI-AuNPを除いて、37°Cで24時間まで互換性がありました。 )37°Cで0時間の場合と比較して、37°Cで24時間の場合(それぞれ0.62と1.0)(表1)。すべての裸およびPCAuNPのZ電位値は、37°Cで0時間の場合と比較して、37°Cで24時間の場合に比較的減少しました。 PBSおよび完全なEMEMでの40および80nm PC BPEI-AuNPの凝集が観察されました。これは、サイズ分布の複数のピークおよびD H の変化と相関していました。 裸のBPEI-AuNPと比較した吸光度スペクトルの赤方偏移(図1および追加ファイル1:図S1、表1)。これらの結果は、40および80nmのPCとヒト血清アルブミンコロナでコーティングされたBPEI-AuNPがPBSおよびさまざまな細胞培養培地で凝集したという最近の研究によって裏付けられました[7,8,9]。
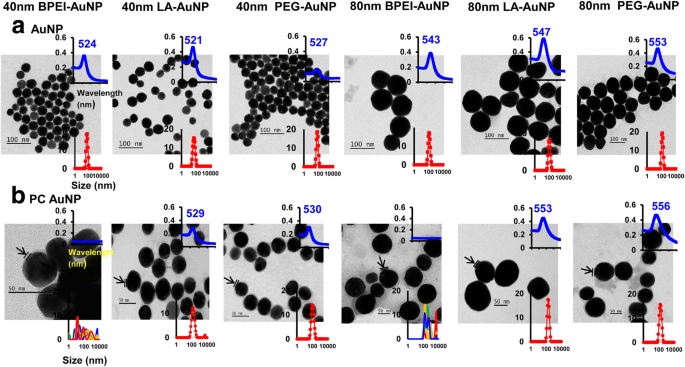
a の透過型電子顕微鏡写真 脱イオン水中のAuNPおよび b 25°Cで0時間のPBS中のPCAuNP、UV-Visスペクトル波長(上の挿入図)、および動的光散乱分布(下の挿入図)。矢印はPCの形成を示します。 PC ヒト血漿タンパク質コロナ、 BPEI 分岐ポリエチレンイミン、 LA リポ酸、 PEG ポリエチレングリコール
AuNP細胞毒性
AuNP細胞毒性は、半数致死濃度(LC 50 )を使用して測定されました。 )C3Aセルで。 NP依存LC 50 周辺のNP表面電荷、粒子サイズ、およびPC形成 AuNPを使用した分析を図2に示しました。すべての40nm BPEI-、LA、およびPEG-AuNPと80 nm BPEI-AuNPは、対応するLC 50 を使用してC3A細胞に対して細胞毒性を示しました。 範囲は127.3〜205.5μg / cm 2 (図2a)。 80 nmの裸のPEG-AuNPは、250μg/ cm 2 の最高濃度で59%の細胞生存率を示しました。 、一方、80nmのLA-AuNPは細胞毒性がありませんでした。 PCは、250μg/ cm 2 で51%の細胞生存率を示した80 nm BPEI-AuNPを除いて、サイズと表面電荷の変化の関数としてAuNP毒性を低減しました。 24時間で(図2b)。最近の研究では、40 nmの裸のBPEI-AuNPは、初代ヒト肝細胞、HUVEC、およびヒト腎近位尿細管細胞(HPTC)に対して毒性があることが示されました(LC 50 範囲は22.4〜80.3μg / cm 2 )[7,8,9]。 PCでコーティングされたBPEI-AuNPはヒト肝細胞に対して細胞毒性を示しましたが、HSAでコーティングされたAuNPは細胞毒性を示しませんでした[7]。これらの結果は、C3A細胞は、癌細胞株の高い増殖率と代謝活性のために、初代ヒト細胞よりもAuNP毒性に対して耐性があることを示唆しています[31]。
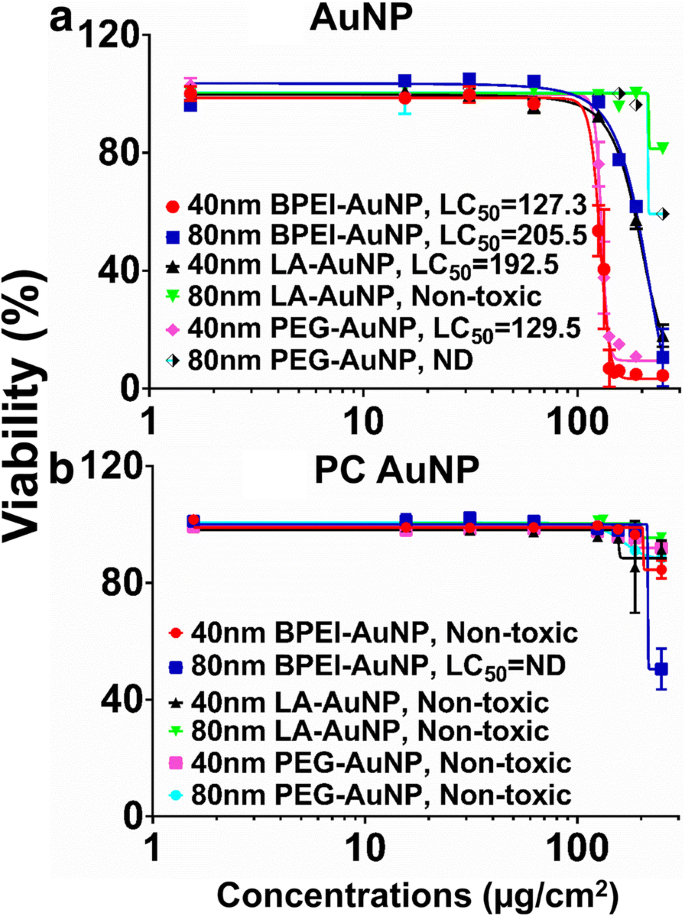
C3Aの実行可能性とLC 50 40および80nmの値 a AuNPと b PCAuNP。データは平均±S.Dを表します。 ( n =3)。 PCヒト血漿タンパク質コロナ、ND未定、BPEI分岐ポリエチレンイミン、LAリポ酸、PEGポリエチレングリコール、LC 50 半数致死量
AuNPの細胞内取り込み
すべての裸およびPCAuNPのNPサイズ、表面電荷、およびPCに依存する細胞取り込みは、1.56μg/ cm 2 と測定されました。 最大24時間。 ANOVAは、サイズ、PC、および時間とともに有意な変化を示しました( p <0.0001)、および相互作用(PC×サイズ、PC×時間、サイズ×時間、およびPC×サイズ×時間)( p <0.001)LA-およびPEG-AuNP取り込み( p )のわずかな相互作用(PC×サイズ)に加えて、すべてのAuNP取り込み =0.2)。図3a–fに示すように、40nmと80nmのベアとPCAuNPの細胞取り込みの直線的な増加が観察されましたが、40nmのベアとPCBPEI-AuNPは、6時間で最高の細胞取り込みに達し、その後減少しました(図3a)。ただし、24時間では、40 nmのカチオン性BPEI-AuNPが最も高い取り込みを示し、次に中性の40 nm PEG-AuNP、次にアニオン性の40 nm LA-AuNPが含まれ、AuNPのC3A細胞の細胞毒性の順序に関連していました(図2a )。この結果は、カチオン性ポリ(N-(2-アミノエチル)アクリルアミド)-およびBPEI-AuNPが、アニオン性ポリ(アクリル酸)-およびLA-AuNPおよび中性ポリ(ヒト結腸直腸腺癌Caco-2細胞、HPTC、およびヒト肝細胞におけるN-(2,3-ジヒドロキシプロピル)アクリルアミド-およびPEG-AuNP [9、32]。さらに、NP-PC複合体は40および80nmのBPEI- LA-AuNPとC3A細胞での40nm PEG-AuNPですが、80 nm PEG-AuNPの取り込みを加速しました(図3f)。これらの結果は、PCがHUVEC、HEK、およびHPTCでAuNPの取り込みを阻害したという最近の研究によって裏付けられています。サイズや表面電荷に関係なく[8、9、33]。対照的に、PCおよびHSAコロナはヒト肝細胞での40 nm PEG-AuNP取り込みを増強しましたが、後者はHEKで80 nmPEG-AuNP取り込みを誘導しました[7、33]。 。
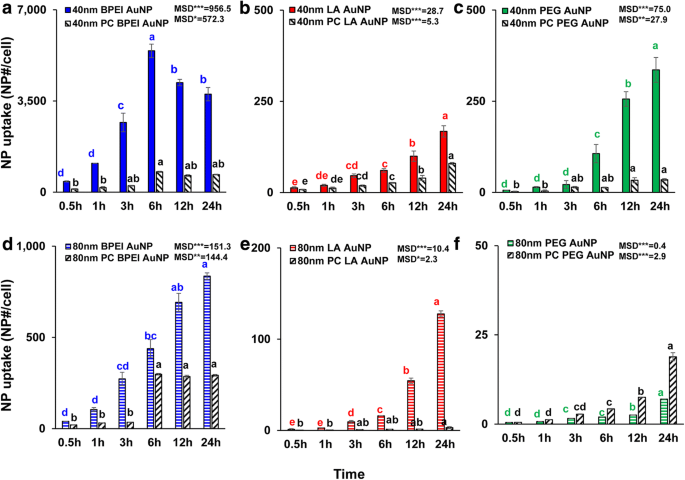
40 nm a の時間依存性細胞取り込み BPEI-AuNP、 b LA-AuNP、および c PEG-AuNP、および80 nm d BPEI-AuNP、 e LA-AuNP、および f 24時間までのC3AセルにPCが存在する場合と存在しない場合のPEG-AuNP。データは平均±S.Dを表します。 ( n =3)。テューキーのHSD検定によると、文字は大幅に異なっていました。 BPEI 分岐ポリエチレンイミン、 LA リポ酸、 PEG ポリエチレングリコール、 PC ヒト血漿タンパク質コロナ、 MSD 最小の有意差。 * p <0.05; ** p <0.005; *** p <0.0001
酸化ストレスおよびニトロソ化ストレスの測定
40 nmの裸のBPEI-およびPEG-AuNPは、他のAuNPと比較してC3A細胞で高い細胞毒性と細胞取り込みを示したため、AuNPによって誘発される酸化/ニトロソ化ストレスを調査するために選択されました。両方のAuNPは、時間および濃度に依存してC3A細胞のROS / RNS生成を調節しました( p <0.0001)および相互作用による(時間×濃度、 p <0.0001)。図4aに示すように、ROS / RNSの生成は、40 nm BPEI-AuNPの高濃度(62.5μg/ cm 2 )で減少しました。 および125μg/ cm 2 )37°Cで1時間ですが、24時間まで増加します。対照的に、40 nm PEG-AuNPは、39.1μg/ cm 2 でROS / RNS生成を大幅に抑制しました。 倍率変化<0.5から24時間まで(図4b)。細胞死の活性化はしばしばNP毒性に寄与し、ほとんどの場合、酸化ストレスにつながるROS / RNS産生の増加がNP毒性の原因となります[34]。表面電荷に依存するROS / RNSの生成は、40nmのカチオン性BPEIおよびニュートラルPEG-AuNPで観察されました。 40 nm BPEI-AuNPは、高濃度でROS / RNS生成の二相性パターン(1時間で抗酸化剤、3時間以降で酸化促進剤)を示しました。これは、C3A細胞での細胞毒性と関連していました(図2a)。この結果は、40nmおよび80nmのBPEI-AuNPおよび20nmのクエン酸-AuNPによって誘導されるROS生成が、時間および濃度に依存して、それぞれヒト肝細胞およびHepG2細胞における細胞毒性と関連していたという以前の研究と一致しています。 [7、35]。 AuNPは、サイズに関係なく、ヒト前骨髄球性白血病細胞であるHL-60で酸化ストレスによる細胞毒性を示し、グルタチオンが完全に減少しました[19]。対照的に、40 nm PEG-AuNPは抗酸化剤として機能し、酸化/ニトロソ化ストレスがC3A細胞における40 nm PEG-AuNP誘発細胞毒性の直接的なメカニズムではない可能性があることを示唆しています(図2b)。
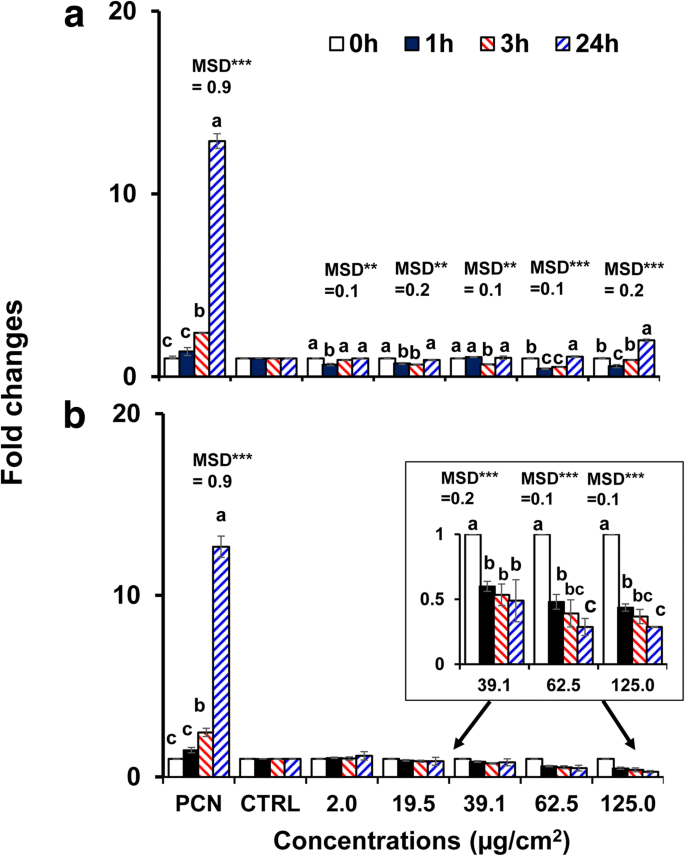
a に曝露されたC3A細胞における時間および濃度依存性のROS / RNS産生 40 nmBPEI-AuNPおよび b 最大24時間の40nmPEG-AuNP。データは平均±S.Dを表します。 ( n =3)。テューキーのHSD検定によると、文字は大幅に異なっていました。 BPEI 分岐ポリエチレンイミン、 LA リポ酸、 PEG ポリエチレングリコール、 CTRL コントロール、 MSD 最小有意差、 PCN ピオシアニン(ROSインデューサー)。 ** p <0.005; *** p <0.0001
CYP3A4アクティビティ測定
CYP3A4活性に対する40および80nmのベアおよびPCAuNPの阻害効果が特徴づけられました。図5に示すように、LC 50 での40nm BPEI-、LA-、およびPEG-AuNPと80 nmBPEI-AuNP 値は、C3A細胞におけるCYP3A4の触媒活性を阻害し、サイズと表面電荷に関係なく、対照と比較して20.1〜31.4%の対応する活性を示しました。無毒濃度の80nm LA-およびPEG-AuNPも、その活性を抑制しました(それぞれ、31.4%および26.6%)。ただし、PCは、40および80 nmのPEG-AuNPに加えて、40および80 nmのAuNPによって誘発されるCYP3A4阻害を大幅に改善し、コントロールと比較して63および46%の活性を示しました。これは、アニオン性タンニン酸-AuNPとカチオン性40および80 nmBPEI-AuNPがCYP3A4の触媒活性を実質的に阻害したというヒト肝臓組織および肝細胞を用いたinvitro研究と一致しています[7、25]。対照的に、カチオン性PEI-AuNPおよび中性ポリビニルピロリドン-AuNPは、HepG2細胞におけるCYP1A2、CYP2C9、およびCYP3A4のmRNA発現、およびラット肝臓スライスにおけるCYP2BおよびCYP3Aをそれぞれ誘導しました[36、37]。最近の研究によると、40および80nmの裸およびPCBPEI-AuNPは、タンパク質のコンフォメーション変化または可逆的阻害としての基質ポケットの遮断を介して、ヒト肝細胞のCYP3A4活性を実質的に抑制しました[7]。
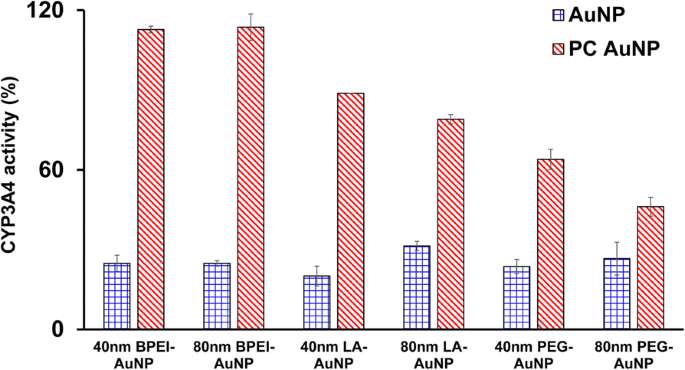
PCの非存在下および存在下で24時間40および80nm BPEI-、LA-、およびPEG-AuNPに曝露されたC3A細胞のCYP3A4活性に対するAuNPの阻害効果。値は平均±S.Dを表します。 ( n =3)。 BPEI 分岐ポリエチレンイミン、 LA リポ酸、 PEG ポリエチレングリコール、 PC ヒト血漿タンパク質コロナ
40 nmPEG-AuNPの毒性経路に焦点を当てた遺伝子発現プロファイリング
13の異なるストレスおよび毒性経路をカバーする代表的な遺伝子から、合計212の遺伝子(↓186および↑26の遺伝子)がLC 50 で差次的に発現されました。 40 nm PEG-AuNPの値(図6、追加ファイル1:表S2〜S7)。全遺伝子(212遺伝子)の12.3%(26遺伝子、↓26、↑0遺伝子)は主にミトコンドリアの脂肪酸β酸化に関与していた。アポトーシスの場合11.3%(24遺伝子、↓18、↑6遺伝子); DNA損傷および修復経路の場合11.3%(24遺伝子、↓18、↑6遺伝子);熱ショック応答については11.3%(24遺伝子、↓22、↑2)。
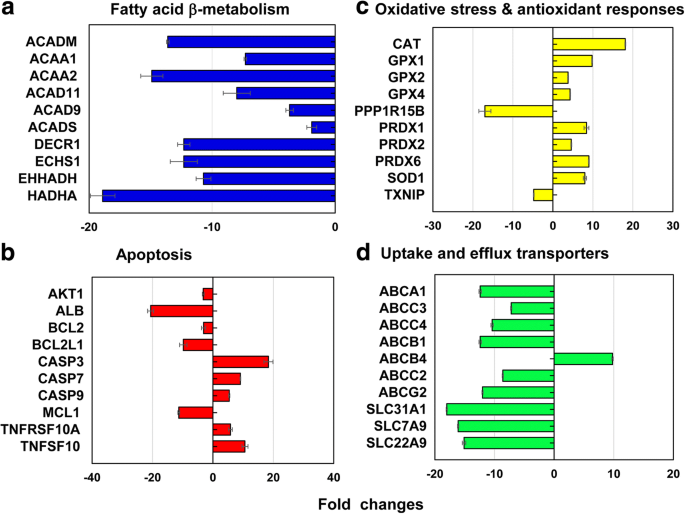
a に関与する代表的な遺伝子 ミトコンドリアの脂肪酸β酸化、 b アポトーシス、 c 酸化ストレスと抗酸化反応、および d LC 50 での肝取り込みおよび排出トランスポーター 40 nmPEG-AuNPの値。すべてのデータは、 p で倍率変化<−2および> 2を示しました。 <0.05。遺伝子オントロジー分析は、追加ファイル1:表S2〜S7
にリストされています。ミトコンドリアの脂肪酸β酸化経路では、アシルCoAの生成に関与し、NADHとFADHの同等物を還元する3つの異なる酵素をコードする遺伝子 2 主に抑制されました。アシルCoAデヒドロゲナーゼのACAD11、ACAD9、ACADM、およびACADS遺伝子(2.0〜13.6倍)。ケトアシル-CoAチオラーゼ中のACAA1およびACAA2(7.3〜14.9倍);エノイルCoAヒドラターゼ中のDECR1、ECHS1、EHHADH、およびHADHA(10.7〜18.9倍)(図6a、追加ファイル1:表S2)。ミトコンドリアの脂肪酸β酸化は、アシルCoAの生成と、NADHおよびFADH 2 の当量の減少に重要な役割を果たします。 、4つの主要な酵素(アシル-CoAデヒドロゲナーゼ、エノイル-CoAヒドラターゼ、ヒドロキシアシル-CoAデヒドロゲナーゼ、およびケトアシル-CoAチオラーゼ[38、39]。さらに、電子担体、NADHおよびFADH 2 > 、トリカルボン酸(TCA)回路とミトコンドリア呼吸鎖に関与し、ATP産生をもたらします。現在の研究では、40 nm PEGはミトコンドリア機能障害を誘発し、ATPおよびFADH 2 の細胞内レベルの低下を介してATP維持の喪失を引き起こしました。 その結果、C3細胞におけるその細胞毒性を定義します(図2a)。同様の現象が、40 nm BPEI-AuNPに曝露されたヒト肝細胞、HUVECおよびHPTCで報告され、ミトコンドリア機能障害が表面電荷や細胞型に関係なく、AuNP毒性の一般的なメカニズムである可能性があることを示しています[7,8,9]。最近の研究では、STAT3リン酸化の阻害剤であるOPB-51602に応答して、不死化した前立腺癌上皮細胞と肺癌上皮細胞でミトコンドリア機能障害関連の細胞毒性が観察されたことが報告されています[40]。
アポトーシス経路では、CASP3、CASP7、CASP9、TNFRSF10A、TNFRSF10B、およびTNFSF10の6つのアポトーシス促進遺伝子がアップレギュレートされたのに対し、AKT1、ALB、BCL2、BCL2L1、MCL1、およびXIAPの6つの抗アポトーシス遺伝子はダウンレギュレートされました(図.6b、追加ファイル1:表S3)、これはC3A細胞の用量依存性細胞毒性と相関していた(図2a)。 DNA損傷および修復チェックポイントでは、チェックポイントキナーゼ(CHEK1 / 2)、DNA除去修復遺伝子(ERCC1 / 2/3)、およびDNAリガーゼIV(LIG4)の遺伝子がアップレギュレートされましたが、他の除去修復遺伝子(ERCC5 / 6、XRCC1 / 5)、チェックポイントキナーゼ(CDKN1A)、およびプロテインキナーゼ(PRKDC)遺伝子がダウンレギュレートされました(2〜19倍)。これらの結果は、40 nm PEG-AuNPによる細胞周期およびDNA修復システムへの干渉が、C3A細胞における細胞死の誘導と相関している可能性があることを示唆しています(図2a、追加ファイル1:表S3)。 2つの異なる熱ショックタンパク質(HSP)(A1AおよびA1B)をコードする遺伝子はアップレギュレーションされました(10.2〜14.2倍)が、HSP40サブファミリーA、B、およびC。 HSP90メンバー1;およびHSP60はダウンレギュレーションされました(2〜16倍)(追加ファイル1:表S4)。
酸化ストレスと抗酸化反応では、LC 50 の40nmPEG-AuNP 値は抗酸化遺伝子を誘導し、酸化促進剤を抑制しました。これは、抗酸化物質自体であるROS / RNS生成の減少に関連していました(図4b)。抗酸化遺伝子では、グルタチオンペルオキシダーゼ(GPX)1、GPX2、GPX4、PRDX1、PRDX2、PRDX6、スーパーオキシドジスムターゼ(SOD)1、およびCATが誘導されました(3.8〜18.1倍)。酸化促進遺伝子では、TXNIPとPPP1R15Bが抑制されました(それぞれ4.8倍と17倍)(図6c、追加ファイル1:表S5)。これは、AuNPがサイズに関係なくHepG2およびヒト肝細胞で酸化ストレス誘発性細胞毒性を示したという以前の研究と一致しています[7、19]。
フェーズI代謝では、CYP3A4およびESD遺伝子が大幅に抑制されました(それぞれ7倍および12倍)。特に、CYP3A4発現に対する40 nm PEG-AuNPの阻害効果は、CYP3A4活性の低下と相関していました(図5)。最近の研究によると、40 nm BPEI-AuNPは、ヒト肝細胞におけるCYP1A2、CYP2C9、およびCYP3A4の遺伝子発現を阻害しました。 HUVECのESD; HPTCのCYP1A1 [7,8,9]。疫学研究は、HCC患者の肝臓組織におけるCYP酵素が、分子レベルおよび機能レベルで腫瘍形成プロセスによって実質的に阻害されることを示しました[41]。
薬物の取り込みと排出トランスポーターの遺伝子発現プロファイリング
腫瘍細胞による多剤耐性(MDR)の発症は、がん治療の失敗の主な原因の1つです[42、43]。内在性膜輸送体を介した薬物取り込みの減少と、P糖タンパク質(P-gp)や乳癌耐性タンパク質(BCRP)などの薬物排出の増加は、MDRの主要なメカニズムの1つです。
40 nm PEG-AuNPに曝露されたC3A細胞における薬物排出および取り込みトランスポーターの差次的遺伝子発現は、ABCトランスポーターの合計14遺伝子(↓12および↑2遺伝子)およびSLCトランスポーターの合計21遺伝子(↓21および↑0遺伝子)はLC 50 で実質的に調節されました 値(図6dおよび7、追加ファイル1:表S7)。 ABCファミリーの薬物排出トランスポーターでは、多剤耐性関連タンパク質(MRP3 / ABCC3)、MRP4(ABCC4)、および基底外側膜のコレステロール流出調節タンパク質(CERP / ABCA1)の遺伝子がダウンレギュレーションされました(7.2〜10.4倍)。小管排出トランスポーターのP-gp(ABCB1)、MRP2(ABCC2)、BCRP(ABCG2)、およびステロリン2(ABCG8)をコードする遺伝子も抑制されました(8.6〜13.8倍)。対照的に、小管膜の多剤耐性(MDR4 / ABCB4)とミトコンドリア外膜のミトコンドリアABCトランスポーター(MTABC3 / ABCB6)は高度にアップレギュレートされていました(それぞれ9.8倍と5.8倍)。薬物取り込みトランスポーターでは、銅トランスポータータンパク質(CTR1 / SLC31A1)および程度は低いが有機アニオン輸送(OAT7 / SLC22A9)の遺伝子も阻害された(それぞれ18倍および15倍)。これらの結果は、40 nm BPEI-AuNPがヒト肝細胞のMDR3をダウンレギュレートするが、HUVECのMRP3をアップレギュレートするという最近の研究を裏付けており、AuNPと排出トランスポーター間の表面電荷および細胞型依存性相互作用を示しています[7、8]。疫学研究は、BCRPの高発現とOCT3の低発現がHCC腫瘍で発生したことを示しました。これは腫瘍の進行とそのサイズと密接に関連していました[44]。以前の研究では、P-gp阻害剤であるベラパミルが、細胞内薬物濃度を増加させることにより、ネコ線維肉腫細胞株におけるドキソルビシンと結合したグルタチオン-AuNPの細胞毒性を増強することが示されました[45]。現在の研究では、40 nm PEG-AuNPに関するメカニズムから得られた情報により、ミトコンドリアの脂肪酸β酸化、TCA回路と呼吸鎖、薬物排出と取り込みトランスポーター、およびCYP3A4活性に対する別個の、しかし依然として補完的な作用が特定されたことが強調されています。 C3A細胞(図7)。最後に、これにより、AuNPと主要な生物学的プロセスとの相互作用およびHCCにおけるその基礎となる分子メカニズムが浮き彫りになり、HCC治療におけるより効果的な治療標的の開発にさらに関与する可能性があります。
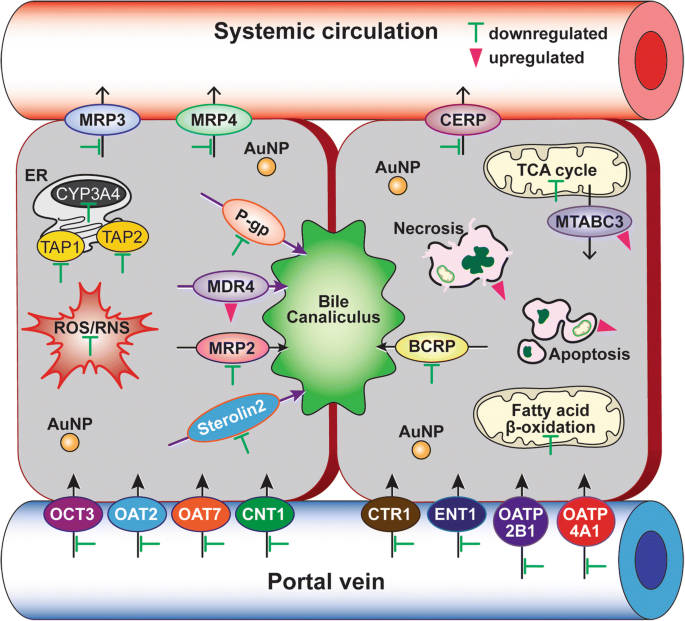
HCC治療における40nmPEG-AuNPの基本的な作用機序の概略図。緑のバー(抑制)とピンクの三角形(誘導)は、40nmのPEG-AuNPで修飾された生物学的マーカーと経路を示しています。遺伝子オントロジー分析は、追加ファイル1:表S2〜S7
にリストされています。RT 2 からの遺伝子発現解析を検証するには アレイでは、9つの遺伝子がリアルタイムPCR用に選択されました。追加ファイル1:表S1では、9つの遺伝子すべてがLC 50 で変調されました。 40 nmPEG-AuNPのこれらの転写変化は、PCRアレイを用いた遺伝子発現解析の変化と一致していました(図6、追加ファイル1:表S2〜S7)。
結論
カチオン性BPEI-、アニオン性LA-、または中性PEG-AuNPとヒト血漿タンパク質コロナ(PC)との相互作用が、 D の変化を引き起こしたことを示しました。 H 、PDI、およびAuNPのz電位、さらにC3A細胞の細胞応答に影響を与えました。すべての裸(PCなし)の40および80 nm AuNPは、80 nm LA-AuNP以外のC3A細胞に対して細胞毒性がありましたが、PCは80 nmBPEI-AuNP以外の細胞毒性を完全に改善しました。 40 nmの裸のBPEI-AuNPが最も高い細胞取り込みを示し、次に40 nm PEG-AuNP、次に40 nm LA-AuNPが続きましたが、PCは80 nmPEG-AuNP以外にAuNP取り込みを抑制しました。 40 nm BPEI-AuNPは、C3A細胞で酸化ストレス(抗酸化剤および抗酸化剤)の二相性応答を引き起こしましたが、40 nmPEG-AuNPは抗酸化剤でした。 CYP3A4活性は、サイズや表面電荷に関係なく、すべての裸のAuNPによって大幅に抑制されましたが、PCは、40および80nmのPEG-AuNP以外の酵素活性に対する阻害効果を大幅に改善しました。 LC 50 で差次的に発現する遺伝子 40 nm PEG-AuNPの値は、主にミトコンドリアの脂肪酸のβ酸化と、程度は低いものの肝排出/取り込みトランスポーターに関与していました。 40 nm PEG-AuNPは、β酸化の3つの主要な酵素(アシルCoAデヒドロゲナーゼ、エノイルCoAヒドラターゼ、ケトアシルCoAチオラーゼ)、TCA回路の他の酵素、およびATP生成のためのミトコンドリア呼吸鎖を阻害しました。 40 nm PEG-AuNPは、LC 50 でアポトーシス促進遺伝子の発現を増加させ、抗アポトーシス遺伝子を減少させました。 価値。 40 nm PEG-AuNPに曝露されたC3A細胞では、高レベルの抗酸化物質と低レベルの酸化促進遺伝子が観察されました。さらに、基底外側膜と小管膜の両方に位置する薬物排出および取り込みトランスポーターの遺伝子が実質的に調節された。
略語
- ANOVA:
-
一元配置分散分析
- AuNP:
-
裸の金ナノ粒子:PCなし
- BPEI:
-
分岐ポリエチレンイミン
- CYP:
-
シトクロムP450
- D H :
-
流体力学的直径
- DLS:
-
動的光散乱
- EDTA:
-
エチレンジアミン四酢酸
- EMEM:
-
イーグル最小必須培地
- HCC:
-
ヒト肝細胞癌
- HPTC:
-
ヒト腎近位尿細管細胞
- HSD:
-
テューキーの正直な有意差検定
- HUVEC:
-
ヒト臍帯静脈細胞
- ICP-MS:
-
誘導結合プラズマ質量分析
- LA:
-
アニオン性リポ酸
- LC 50 :
-
半数致死量
- MDR:
-
多剤耐性
- NP:
-
ナノ粒子
- PBS:
-
リン酸緩衝生理食塩水
- PC:
-
ヒト血漿タンパク質コロナ
- PDI:
-
多分散度指数
- PEG:
-
中性ポリエチレングリコール
- RNAi:
-
RNA干渉
- RNS:
-
活性窒素種
- ROS:
-
活性酸素種
- SO:
-
スーパーオキシド
- TEM:
-
透過型電子顕微鏡
- TFF:
-
タンジェンシャルフローろ過
ナノマテリアル
- ホテルのメンテナンスとその収益への影響
- 局在表面プラズモン共鳴に基づく金ナノバイオセンサーは、ヒトブルセラ症を診断することができ、迅速で手頃な方法を導入します
- 1D混合二元酸化物CeO2-LaOx担持金触媒の合成とCO酸化活性
- ポリ(3,4-エチレンジオキシチオフェン)/金/グラフェン複合材料の固体加熱合成とその亜硝酸塩およびヨウ素酸塩のアンペロメトリー定量への応用
- 6-メルカプトプリンとニューロン透過性ペプチドで修飾された金ナノ粒子によるSH-SY5Y細胞増殖の促進
- 磁性金ナノ粒子標識ヘパラナーゼモノクローナル抗体とその後の腫瘍磁気共鳴画像法への応用
- インビボでのDNAとのジフェニル-N-(トリクロロアセチル)-アミドホスフェート相互作用に対するC60フラーレンの効果およびinvitroでのヒト白血病細胞株に対するその細胞毒性活性
- PEG-PCCLナノ粒子の毒性評価とパクリタキセル負荷の抗腫瘍効果に関する予備調査
- カーボンナノチューブとその誘導体がinvitroでの腫瘍細胞および生化学的パラメーター、invivoでの細胞血液組成に及ぼす影響
- 金ナノ粒子で阻害されたチトクロームP4503A4活性と、ヒト肝細胞癌細胞株C3Aにおけるその細胞毒性の根底にある分子メカニズムの評価
- 蛍光ネオ糖タンパク質金ナノクラスター:合成と植物レクチンセンシングおよび細胞イメージングへの応用