


反強磁性MnBr2から大きなMAEを持つ強磁性Mn3Br8単分子層への会話
要約
低エネルギースピントロニクスの差し迫った必要性は、キュリー温度が液体窒素温度(77 K)を超え、かなりの磁気異方性を持つ2次元(2D)強磁性体です。 Mn 3 を研究しました Br 8 MnBr 2 の人口1/4でMn空孔を誘導することによって得られる単分子層 単層。このような欠陥のある構成は、Mn-d 5 の配位構造を変更するように設計されています。 かなりの磁気異方性エネルギー(MAE)で強磁性を実現します。私たちの計算によると、Mn 3 Br 8 単分子層は、キュリー温度130 K、式単位あたり-2.33 meVの大きなMAE、13 /3μ B の原子磁気モーメントを持つ強磁性(FM)ハーフメタルです。 Mn原子の場合。 さらに、Mn 3 Br 8 単分子層は、5%の圧縮ひずみ下でのキュリー温度が160 Kである小さな二軸ひずみ下でFMを維持します。さらに、二軸ひずみとキャリアドーピングの両方により、MAEが増加します。これは、主に磁気結晶異方性エネルギー(MCE)によってもたらされます。 MnBr 2 の設計された欠陥構造 単分子層は、2D材料で大きなMAEを使用して強磁性を実現するためのシンプルで効果的な方法を提供します。
はじめに
スピントロニクスは、電子スピンとそれに関連する磁気モーメントを利用しており、電荷ベースのデバイスに勝る独自の利点があるため、過去数十年の間に大きな注目を集めてきました[1]。有限温度での長距離磁気秩序を備えた2次元(2D)強磁性体の最近の実現[2、3]は、ナノスケールのスピントロニクスおよび関連するアプリケーションにとって非常に重要であり、2D強磁性体の調査と製造に多大な努力を促します[4,5 、6,7,8,9]。
原子の厚さを持つ最初の2つの2D強磁性体、つまり単層CrI 3 が2017年に達成されました。 [2]および二重層Cr 2 Ge 2 Te 6 [3]。残念ながら、両方のキュリー温度は液体窒素温度(77 K)よりも低いため、実際のアプリケーションは制限されます。キュリー温度に加えて、かなりの磁気異方性と磁気モーメントも実用化に欠かせません。大きな磁気異方性エネルギー(MAE)は、熱変動に対する磁気秩序の利点と、情報のビットあたりの粒子サイズを縮小する可能性を意味します。 MAEが小さいと、強磁性ではなく超常磁性になる可能性があります。大きな磁気モーメントは、スピントロニクスの感度、効率、密度を高めます。重元素は、その強いスピン軌道相互作用(SOC)効果により、大きなMAEをもたらす可能性が高くなります[10]。 CrI 3 のように、重元素で構成される一連の2D FM材料は、MAEが大きいと予測されています。 [11]、CrAs [12]、CrSeI [13]、CrSiTe 3 [14]、CrWI 6 [15]、FeBr 2 およびFeI 2 単層[16]。さらに、MXenes Mn 2 のMn原子の局所磁気モーメント NF 2 およびMn 2 N(OH) 2 は4.5μ B 報告されているFM2D材料の中で最大のMn原子あたり[17]。
CrI 3 以降 単分子層の合成に成功し、遷移金属ハロゲン化物が多くの注目を集めています[18、19、20、21、22、23、24、25、26、27]。スピンゼーベック効果は、二重層MnF 2 で観察されています。 [20]; CrI 3 の数層 磁気トンネリングジャンクション(MTJ)に実装されています[21]。 NiCl 3 単分子層は、新しいディラックスピンギャップレス半導体(SGS)であると予測されています[22]。特に、MnBr 2 単分子層は反強磁性であり、第一原理計算に基づいて、平面に垂直な方向に沿って0.25 meVMAEです[16]。 Mn 2+ イオンはd 5 にあります 磁気モーメントが5μ B の高スピン状態 [16、26]。これらの結果は、MnBr 2 の可能性を示唆しています。 磁気モーメントの大きい単層強磁性体として。重要な問題は、Mnイオン間のAFM結合をFM結合に変換する方法です。
LaMnO 3 では、有意な密度のMn空孔が実験的に観察されました。 薄膜[28]であり、高エネルギー粒子の照射または化学エッチング[29]を介して合成プロセスを意図的に調整することにより、欠陥の濃度を制御できます。これに関連して、Mn 3 を設計しました Br 8 MnBr 2 に単一のMn空孔を誘導することによる単分子層 単層。空孔はMn原子の配位構造を変化させ、d 5 を破壊します。 反強磁性結合を強磁性結合に変換し、Br原子が重いために大きなMAEをもたらす可能性のある構成。予想通り、Mn 3 Br 8 単分子層はFMであり、式単位あたり-2.33 meVの大きなMAEを持ち、各Mn原子の磁気モーメントは13 /3μ B です。 。柔軟な基板を曲げる[30,31,32,33]、弾性基板を伸ばす[33,34,35]、熱膨張の不一致を利用する[33、36]など[33]、静電ドーピングによるスピン偏極の効果的な制御[37、38]、Mn 3 も研究しました。 Br 8 二軸ひずみおよびキャリアドーピング下の単分子層。私たちの結果は、Mn 3 Br 8 単分子層はFMであり続け、小さな二軸ひずみの下でキュリー温度が上昇します。さらに、二軸ひずみとキャリアドーピングの両方により、MAEが増加する可能性があります。
計算方法
本研究のすべての計算は、ウィーンの ab-initio で実装されているスピン偏極密度汎関数理論(DFT)法を採用して実行されました。 シミュレーションパッケージ(VASP)[39]。電子と原子核の間の相互作用は、プロジェクター拡張波(PAW)法[40、41]によって記述され、電子交換-相関相互作用は、一般化勾配近似(GGA)内で機能するPerdew-Burke-Ernzerhof(PBE)によって記述されました。メソッド[42]。ハバードU項は、強い相関のある相互作用を計算するために採用されました[43]。 Mn-d電子には、Mnを組み込んだ2D材料の研究に採用された4 eVの有効なオンサイトクーロン相互作用パラメータ(U)と1 eVの交換エネルギー(J)が使用されました[44]。ブリュアンゾーン統合は、Monkhorst-Packスキームに基づく9×9×1kメッシュを採用することによって実行されました[45]。フォノンスペクトルは、VASPパッケージ内に実装されているフォノピーコード[46]を使用して計算されました。隣接する層間の相互作用を避けるために、単分子層の表面に垂直な方向に沿って20Åの真空空間が追加されました。平面波基底関数系のカットオフエネルギーは500eVに設定されました。総エネルギーと力の収束基準は1×10 –6 に設定されました。 それぞれeVと0.01eV /Å。
結果と考察
MnBr 2 の劈開エネルギー、基底状態、および安定性 単層
バルクMnBr 2 の最適化された格子定数 はa =b =3.95Åであり、以前の実験結果と一致しています( a = b =3.87Å)[25]。まず、MnBr 2 の剥離の実現可能性を調査しました。 バルクMnBr 2 からの単分子層 。図1aは、劈開エネルギーを計算するための、よく知られた、効果的で、広く承認されている方法を示しています[47、48、49]。具体的には、劈開エネルギーは、図1bに示すように、2つの破壊部分間の分離距離\(d \)に対する基底状態の全エネルギーの変化を計算することによって得られました。aとbの格子定数は次のとおりです。バルクMnBr 2 の平衡状態での値として固定 。層間の長距離vdW相互作用は、GrimmeのDFT-D2スキームによって記述されました[50、51]。図1bに示すように、総エネルギーは分離距離とともに増加し、その後ゆっくりと収束します。計算された劈開エネルギーは0.10J / m 2 、これは、グラファイトの2つの破壊部分間の劈開エネルギー(0.35 J / m 2 )と比較して小さいです。 )[52]、MnBr 2 を取得する可能性を示しています マイクロメカニカル剥離法による単層。
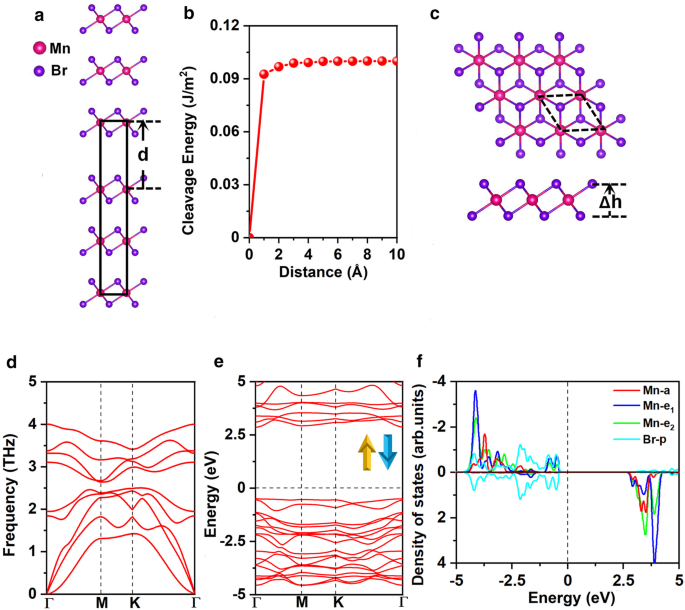
a MnBr 2 のバルクモデル 劈開エネルギーと b の計算に使用されます 2つの破砕部分間の分離距離\(d \)の関数としての劈開エネルギー(平衡層間距離は0に設定されます)。 c 上面図と側面図、 d フォノンスペクトル、 e スピンチャネルと f の両方の電子バンド構造 MnBr 2 のMn-d軌道とBr-p軌道の予測状態密度(PDOS) 単層。 Δ h 2つのハロゲン化物平面間の垂直距離を表します。プリミティブセルは黒い破線で循環されます。バンド構造とDOSのフェルミ準位は0eVに設定されています
MnBr 2 単分子層は、図1cに示すように\(C _ {{{3} v}} \)対称性を持っています。各Mn原子は6つの隣接するBr原子に囲まれ、八面体を形成します[MnBr 6 ] 4- ユニット。図2aおよびbに示すように、3つの可能な磁気構成、つまり非磁性(NM)、強磁性(FM)、および反強磁性(AFM)状態が考慮されます。 Mnイオンの高スピン状態と低スピン状態の両方が考慮されます。私たちの結果は、FM状態のMnイオンがd 1 で低スピンであることを示しています。 AFM状態のMnイオンは、d 5 で高スピンになります。 構成。 MnBr 2 の基底状態 単分子層はAFM状態であり、NMおよびFM状態よりも、式単位あたりそれぞれ3.91eVおよび0.72eV安定しています(追加ファイル1:表S1)。 MAEは0.25meVであり、正の値は磁化容易軸が面外方向に沿っていることを示し、前の結果と一致しています[16]。最適化された格子定数は a = b =3.95Å、バルクMnBr 2 の格子定数と同じ 。 Mn-Br結合長は2.73Åで、2つのハロゲン化物面間の垂直距離は3.03Åです。
a の概略図 強磁性および b MnBr 2 の反強磁性構成 単層
MnBr 2 の安定性 単分子層は、形成エネルギー、フォノンスペクトル、および弾性定数を計算することによってさらに調査されました。形成エネルギーは次のように計算されます:
$$ E _ {{{\ text {form}}}} =E _ {{{\ text {MnBr}} _ {{2}}}} --E _ {{{\ text {Mn}}}}-2E _ {{ {\ text {Br}}}} $$ここで、\(E _ {{{\ text {MnBr}} _ {{2}}}} \)はMnBr 2 のエネルギーを表します 単分子層、\(E _ {{{\ text {Mn}}}} \)および\(E _ {{{\ text {Br}}}} \)は、それぞれバルク構造内のMnおよびBr原子のエネルギーです。計算された\(E _ {{{\ text {form}}}} \)は、原子あたり-1.87eVです。負の値は、地層が発熱性であり、MnBr 2 であることを意味します。 単分子層はエネルギー的に有利です。さらに、MnBr 2 の計算されたフォノンスペクトル(図1d) 単分子層は、ブリュアンゾーン全体で負の周波数を示さず、動的に安定していることを示しています。さらに、計算された弾性定数(追加ファイル1:表S2)は、\(C_ {11}> 0 \)、\(C_ {11} C_ {22} --C_ {12)のBorn-Huang基準[53]に準拠しています。 } ^ {2}> 0 \)および\(C_ {66}> 0 \)、MnBr 2 単分子層は機械的に安定しています。計算された面内剛性は26.98J / m 2 、MnPSe 3 の約75% (36 J / m 2 )[49]、およびMoS 2 の15% 単層(180 J / m 2 )[54]。さらに、MnBr 2 単分子層は、MoS 2 と比較して、より高い柔軟性と、より大きな引張ひずみに耐える能力を示します。 単層(11%)[54]。これは、MnBr 2 のイオン結合に起因する可能性があります MoS 2 の共有結合に対する単分子層 単層。弾性定数に関連する変形の分析は、それがその重量に耐えることができることを示しています(SIの詳細を参照してください)。
MnBr 2 の電子バンド構造 単分子層を図1eに示します。これは、MnBr 2 であることを示しています。 単分子層は、3.35eVの直接バンドギャップを持つ半導体です。価電子帯の最大値(VBM)と伝導帯の最小値(CBM)は、どちらも\(\ Gamma \)ポイントにあります。電子構造の洞察を得るために、Mn-dおよびBr-p軌道の予測状態密度(DOS)を図1fに示します。 Mnイオンの5つのd軌道は、\(a(d _ {{z ^ {2}}})\)、\(e_ {1}(d_ {xz} + d_ {yz})\)、および\( e_ {2}(d_ {xy} + d _ {{x ^ {2} --y ^ {2}}})\)\(C _ {{{3} v}} \)対称性に従ってグループ化します。より悪い電荷分析は、各Mn原子が2つの隣接するBr原子に2つの電子を提供することを示唆しています。したがって、1つのスピンチャネル内の5つのd軌道は、Mn 2+ の5つのd電子によって完全に占有されます。 イオン。同様に、2つのMn 2+ ユニットセル内のイオンはd 5 にあります 磁気モーメントが5μ B の高スピン状態 / −5μ B 、Br 1- イオンは4p 6 の低スピン状態にあります 無視できる磁気モーメント-0.02μ B (追加ファイル1:図S1(a))。 Goodenough–Kanamori–Anderson(GKA)の規則によれば、このような構成は常に反強磁性結合を提供します[55]。
Mn 3 の安定性、電子的、および磁気的特性 Br 8 単層
d 5 を破るために、Mn空孔が導入されました。 Mn 2+ の構成 イオン。単一のMn空孔は、MnBr 2 の\(2 \ times 2 \ times 1 \)スーパーセルに導入されます。 Mn 3 を放出する単分子層 Br 8 単層。図3aに示すように、各Mn原子には4つの最も近い隣接するMn原子があり、6つのBr原子に結合して、歪んだ八面体を形成します[MnBr 6 ] 単位。図4に示す5つの磁気状態(NM、FM、FIM、AFM-1、およびAFM-2)を検討しました。私たちの結果は、FM状態が基底状態であり、式単位あたりそれぞれ9.84 eV、32.90 meV、129.85 meV、および97.65meVだけ他の4つよりも安定していることを示しています。最適化された格子定数は3.95Åのままです。 MnBr 2 とは異なります 単層、Mn 3 Br 8 単分子層には2種類のMn-Br結合があります(図3b)。 Mn原子と2つの中央のBr原子(\(d _ {{\ text {Mn-Br1,2}}} \))の間の結合は2.76Åですが、他のMn–Br結合(\(d _ {{\ text {Mn-Br3,4,5,6}}} \))は2.59Åです。 2つのハロゲン化物平面間の垂直距離は3.33Åです。
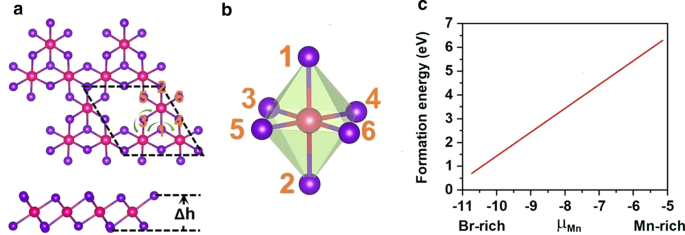
a Mn 3 の上面図と側面図 Br 8 単分子層、\(\ Delta h \)は、2つのハロゲン化物平面間の垂直距離を表します。プリミティブセルは黒い破線で循環されます。緑の矢印の線は、超交換相互作用の2つの異なる経路を示しています。 b 歪んだMnBr 6 の構造 八面体。 c Mnの化学ポテンシャル(μMn)の関数としての単一Mn空孔の形成エネルギー
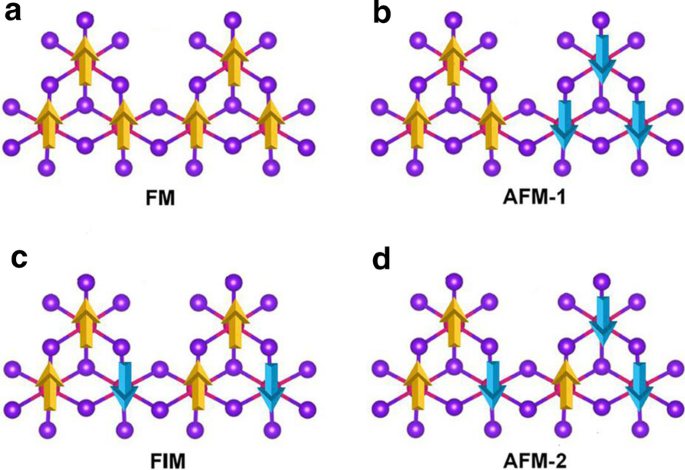
a の概略図 強磁性、 b 反強磁性-1、 c フェリ磁性、および d Mn 3 の反強磁性-2構成 Br 8 単層
Mn空孔を誘発する可能性を検証するために、まず、次の式を使用して、Mnに富む環境とBrに富む環境での空孔形成エネルギーを計算しました。
$$ E _ {{F({\ text {Mn-rich}})}} {\ text {=}} E _ {{{\ text {Mn}} _ {3} {\ text {Br}} _ {8 }}}-(4 \ times E _ {{{\ text {MnBr}} _ {{\ text {2}}}}}-\ mu _ {{{\ text {Mn-max}}}})$$ $$ E _ {{F {\ text {(Br-rich)}}}} {=} E _ {{{\ text {Mn}} _ {{3}} {\ text {Br}} _ {{8} }}}-(4 \ times E _ {{{\ text {MnBr}} _ {{2}}}}-\ mu _ {{\ text {Mn-min}}})$$ここで、\(E _ {{{\ text {Mn}} _ {{3}} {\ text {Br}} _ {{8}}}} \)および\(E _ {{{\ text {MnBr}} _ {{2}}}} \)Mn 3 の総エネルギーを表します Br 8 およびMnBr 2 単分子層\(\ mu _ {{\ text {Mn-max}}} \)は、Mnが豊富な環境下でのMnの化学ポテンシャルであり、バルク構造内のMn原子のエネルギーとして計算されます\(\ mu_ { {\ text {Mn-min}}} \)は、Brが豊富な環境下でのMnの化学ポテンシャルであり、次のように計算されます。
$$ \ mu _ {{\ text {Mn-min}}} =E _ {{{\ text {MnBr}} _ {{2}}}}-2 \ times \ mu _ {{\ text {Br-max}} } $$ここで、\(\ mu _ {{\ text {Br-max}}} \)はBrの化学ポテンシャルであり、気相中のBr原子のエネルギーとして計算されます。図3cに示すように、Mnリッチ/ Brリッチ環境下での形成エネルギーはMn空孔あたり6.30 / 0.71 eVであり、Mn空孔の形成がBrリッチ環境下でエネルギー的に有利であることを示しています。実際、S空孔はMoS 2 で実験的に達成されています。 単分子層[56]であり、Sが豊富な環境下でのS空孔の予測形成エネルギーは2.35 eV [57]です。さらに、β-FeOOH/ PNGN(多孔質窒素ドープグラフェンネットワーク)のような多孔質ナノアーキテクチャを構築すると、かなりのFe空孔を誘発する可能性があり[58]、ブリッジマン法を採用して秩序化Fe空孔を誘発しました。また、これらの方法がMn空孔の誘導に適用できることを期待しています[59]。さらに、Mn 3 のフォノンスペクトルには負の周波数はありません。 Br 8 図5aに示す単分子層で、動的安定性を証明しています。これらの結果は、強磁性をもたらすためにMn空孔を導入するという私たちの設計を承認します。
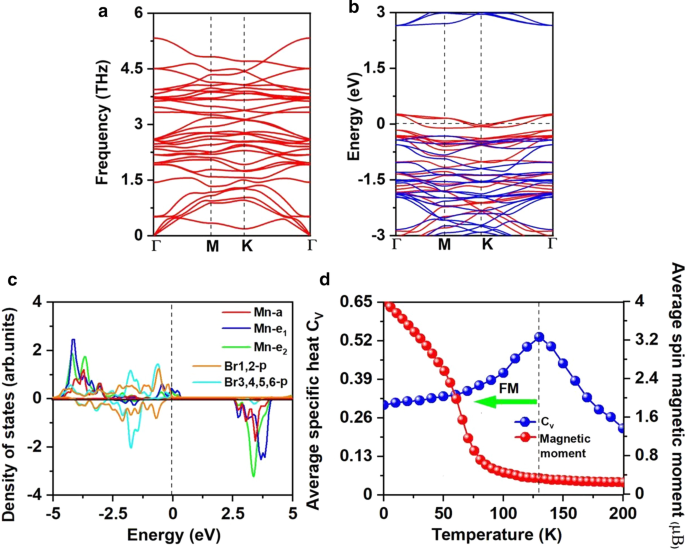
a フォノンスペクトル、 b スピン分解電子バンド構造、および c Mn 3 のMn-d軌道とBr-p軌道の予測状態密度(PDOS) Br 8 単層。 d Mn原子のオンサイト磁気モーメントと比熱 C v Mn 3 のハイゼンベルクモデルに基づく温度の関数として Br 8 単層。バンド構造とPDOSのフェルミ準位は0eVに設定されています
Mn 3 の強磁性 Br 8 FM超交換相互作用に起因する単分子層。 Goodenough–Kanamori–Anderson(GKA)の規則[55]によると、Mn-Br-Mnの角度が約90°の場合、Mnイオン間の超交換相互作用はFMです。このような構成(追加ファイル1:図S2)では、Mn-d軌道は異なる直交Br-p軌道とAFM結合する傾向があるため、間接的なMn-Mn磁気結合はFMであると予想されます。しかし、各MnイオンにMnBr 2 のような5つの不対電子がある場合 単分子層、超交換はAFMですが、MnBr 2 には空のスピンアップMn-d軌道が残っていないため、Mn-Br-Mn角度は90°に近くなります。 単分子層とスピンアップd電子は隣接するMnサイト間をホップできません[60]。 Mn 3 には、既存の2つの異なる超交換相互作用パスがあります。 Br 8 (図3a)、両方ともFMです。 1つは、Mn-Br結合長が2.76ÅでMn-Br-Mn角度が87.5°の中央のBr1,2原子です。もう1つは、Mn-Br結合長が2.59ÅでMn-Br-Mn角度が95°のBr3,4,5,6原子を含みます。図5cに示すように、Br3,4,5,6原子のp軌道とMn-d軌道の間の混成相互作用は、特に-2eVから-1.4eVまで、Br1,2原子を含むp-d混成の相互作用よりも強力です。 1.4から-0.9eVの間、 p - d Br1,2原子が関与する混成軌道が支配的です。
より悪い電荷分析は、各Mn原子が隣接するBr原子に8/3の電子を提供することを示唆しています。したがって、MnイオンはMn 8/3 + にあります。 州。図5cに示すように、各Mnイオンの13/3の電子はすべて、d軌道のスピンアップチャネルを満たしますが、Br 1- イオンは4p 6 の低スピン状態にあります 。したがって、各Mn 8/3 + の磁気モーメント イオンは13 /3μ B ; Br 1- の磁気モーメント イオンは無視できます(追加ファイル1:図S1(b))。空孔による強磁性の誘導は、d 0 でも観察できます。 ZnSやZnO [61、62]のようなシステムでは、単一の空孔が2μ B の磁気モーメントを誘発する可能性があります。 [61] 。 各Mnイオンについて、2/3のd軌道は占有されていません。 \(e_ {1} \)軌道と\(e_ {{2}} \)軌道の両方のスピンアップチャネルが部分的に占有され、フェルミ準位を横切っているため、金属量が半分になります。ハーフメタリック特性は、図5bに示すスピン分解電子バンド構造からも観察できます。スピンアップチャネルは金属製ですが、スピンダウンチャネルは2.97eVの間接バンドギャップで半導体です。 VBM / CBMは、\({\ text {M}} \)/ \(\ Gamma \)ポイントにあります。バンドギャップの値は、MnP(2.86 eV)[63]、MnAs(2.92 eV)[63]、およびNi 2 の値に近い値です。 いいえ 2 (2.98 eV)[64]、これは熱的に励起されたスピンフリップを防ぐのに十分な大きさです。 MnBr 2 との比較 単分子層では、半導体チャネルのVBMとCBMの両方がフェルミ準位に近づきます。 CBMは依然としてMn原子によって支配されていますが、VBMは新しいBr1,2原子によって支配されています。一方、半導体チャネルは直接から間接に変換され、バンドギャップが減少します。同様の現象がMnCl 2 でも観察されました。 H官能化を伴う単分子層[60]。
磁化方向は、磁気異方性エネルギー(MAE)によって決定されます。固体のMAEは、スピン軌道相互作用(SOC)に関連する結晶磁気エネルギー(MCE)と、静磁気双極子-双極子相互作用に起因する磁気双極子異方性エネルギー(MDE)の2つの要因から生じます。 bccFeやfccNiなどの3D等方性材料のMDEは非常に小さいです。しかし、大きな磁気モーメントを持つ遷移金属原子で構成される低次元材料の場合、MDEを無視するべきではありません[65,66,67]。 MCEは、SOCを考慮した、面内(100または010)方向と面外(001)方向に沿った磁化エネルギーの差として定義されます。 MDEは、面内磁化と面外磁化の間の\(E_ {d} \)の差として取得されます。原子リュードベリ単位の\(E_ {d} \)は、[65、66]
で与えられます。 $$ E_ {d} =\ sum \ Limits_ {ij} {\ frac {{2m_ {i} m_ {j}}} {{c ^ {2}}}} M_ {ij} $$ここで、光速\(c =274.072 \)、\(i / j \)はユニットセル内の原子位置ベクトル、\({m} _ {i} / {m} _ {j} \ )は原子の磁気モーメント(μ B )サイト\(i / j \)。磁気双極子マーデルング定数\(M_ {ij} \)は、
を介して計算されます。 $$ M_ {ij} =\ sum \ Limits_ {R} {\ frac {1} {{\ left | {R + i + j} \ right | ^ {3}}}} \ left \ {{1-3 \ left。 {\ frac {{\ left [{(R + i + j)\ cdot \ mathop {m_ {i}} \ Limits ^ {\ wedge}} \ right] ^ {2}}} {{\ left | {R + i + j} \ right | ^ {2}}}} \ right \}} \ right。$$ここで、\(R \)は格子ベクトルです。 2D材料では、すべての\(R \)と\(i \)が面内にあるため、面外磁化の2番目の項はゼロになり、正の\(M_ {ij} \ )、一方、\(M_ {ij} \)は面内磁化に対して負です[67]。したがって、MDEは遷移金属の磁気モーメントに関連し、常に面内磁化を優先します。
Mn 3 の計算されたMCE Br 8 単分子層は-1.90meV /式単位(図6a)であり、バルクFe(0.001 meV /原子)およびNi(0.003 meV /原子)[68]よりもはるかに大きく、Rh上のFe単分子層よりも大きい(111)(原子あたり0.08 meV)[69]、Mn 3 の磁化を示唆している Br 8 単分子層は熱安定性があります。 MCEと方位角の関係は、次の式で表すことができます[70]:
$$ {\ text {MCE}}(\ theta)=A \ cos ^ {2}(\ theta)+ B \ cos ^ {4}(\ theta)$$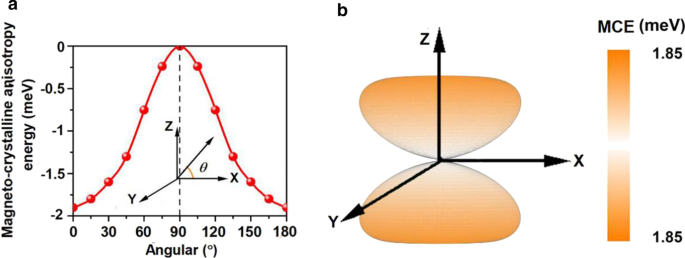
磁気結晶異方性エネルギー(MCE)の変動 a 方位角と b に関して Mn 3 のスペースに Br 8 単層
ここで、\(A \)と\(B \)は異方性定数、\(\ theta \)は方位角です。フィッティングの結果は、追加ファイル1に示されています。 S3。さらに、スピン軸が空間全体を回転するMCEの進化を図6bに示します。 xy平面内のMCEは差がないが、xy平面に垂直な方向に沿って最大値に達し、強い磁気異方性を確認している。 MDEは式単位あたり-0.43meVであり、MAE(MCE + MDE)は式単位あたり-2.33meVです。負の値は、磁化容易軸が面内方向に沿っていることを示します。 MDEは磁化方向を変更しませんが、磁化方向を強化します。さらに、Mn 3 のMAE Br 8 単分子層はMnBr 2 よりもはるかに大きい 単層、私たちのデザインの有効性を再び証明します。
さらに、FM Mn 3 の\(T_ {c} \)を計算しました。 Br 8 ハイゼンベルクモデルに基づくモンテカルロ(MC)シミュレーションを実行することによる単分子層。これは、2D材料の\(T_ {c} \)を予測するための効果的な方法であることが証明されています[11、15、48、58、71、72 、73、74、75、76]。 CrI 3 の推定\(T_ {c} \) 単分子層は42K(追加ファイル1:図S4)[76]であり、実験測定値[2]および以前の計算結果[15、58、71、72、74、76]とよく一致しており、私たちの採用した方法。最も近い隣接(NN)磁気相互作用を含むスピンハミルトニアンは次のように記述されます
$$ H =-\ sum \ Limits_ {i、j} {JM_ {i} M_ {j}} $$ここで、\(J \)はNN磁気交換パラメーター、\(M_ {i / j} \)はMnイオンの磁気モーメントであり、モンテカルロ法に基づくスピン偏極電子の数に近い積分です[71、77 、78]、\(i \)および\(j \)は、MnイオンのNNペアを表します。磁気結合パラメータ\(J \)は、FM状態とAFM状態のエネルギー差を介して次のように計算されます
$$ J {=} \ frac {{E _ {{{\ text {AFM1}}}}-E _ {{{\ text {FM}}}}}} {{16M ^ {2}}} $$NNMnイオンの計算された\(J \)は1.01meVです。正の値は、FM結合が優先されることを示します。
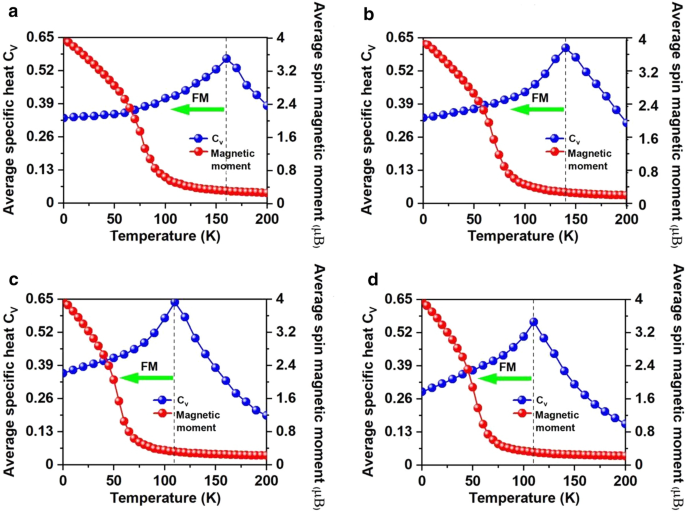
計算されたNNMnイオンの\(J \)と、20,000個の磁気モーメントベクトルを含む\(100 \ times 100 \ times 1 \)スーパーセルを採用して、MCシミュレーションを実行しました。各温度でのシミュレーションは10 5 続きます ステップ。各磁気モーメントベクトルは、すべての方向にランダムに回転します。図5dは、\(C _ {{_ {V}}} ={{\ left({\ left \ langle {E ^ {2}} \ right \ rangle- \ left \ langle E \ right \ rangle ^ {2}} \ right)} \ mathord {\ left / {\ vphantom {{\ left({\ left \ langle {E ^ {2}} \ right \ rangle- \ left \ langle E \ right \ rangle ^ {2}} \ right)} {K_ {B} T ^ {2}}}} \ right。\ kern- \ nulldelimiterspace} {K_ {B} T ^ {2}}} \)温度、ここから、Mn 3 に対して130Kの\(T_ {c} \)を取得しました。 Br 8 液体窒素温度(77 K)よりも高い\(C_ {v} \)のピーク位置、およびCrI 3 の\(T_ {c} \)を特定することによる単分子層 (45 K)[2]およびCr 2 Ge 2 Te 6 (28 K)[3]、CrX 3 (X =F、Cl、Br)(36〜51 K)[11]、CrXTe 3 (X =Si、Ge)(35.7 K、57,2 K)[48]。私たちの計算は、FM Mn 3 Br 8 単分子層は、液体窒素温度よりも高いMAEおよびキュリー温度を持っています。
Mn 3 Br 8 二軸ひずみとキャリアドーピング下の単分子層
ひずみ工学は、多くの2D材料に適用可能であり、結合長や角度などの構造パラメータを変更し、電子的および磁気的特性を調整するのに効果的であることが証明されています。これに関連して、Mn 3 を調査しました。 Br 8 − 5%から5%の範囲の二軸ひずみ下の単分子層。 Mn 3 Br 8 − 5〜5%の二軸ひずみ下の単分子層はFMを維持し、原子磁気モーメントはほとんど変化しません。図1と図2に示すように。 7aおよびc、2つのMn原子とBr1,2原子の間の角度(θ Mn-Br1,2-Mn )は84°–90°で、ひずみが大きくなるにつれて増加し、徐々に90°に近づきます。 Br3,4,5,6原子を含むMn–Br–Mn角度(θ Mn-Br3,4,5,6-Mn )90°から100°の範囲で、90°から徐々にずれます。したがって、異なる直交Br-p軌道を介して媒介されるMnイオン間の超交換相互作用は依然としてFMです。
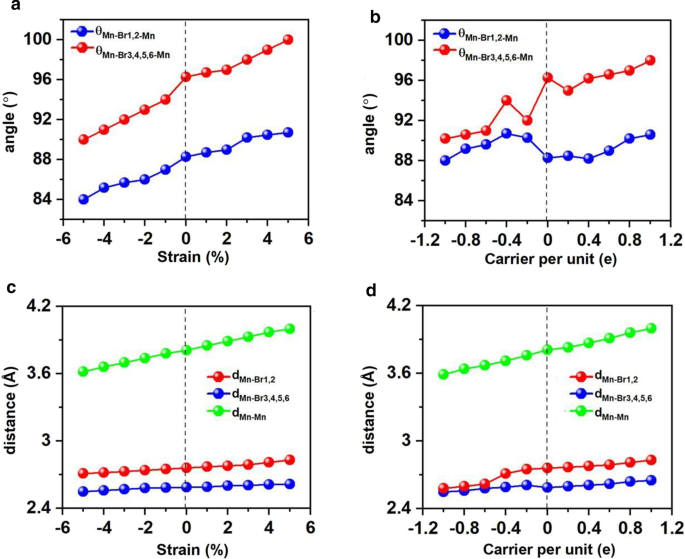
2つのMnとBr原子間の角度の変化、MnとBr原子間の距離、および適用された2軸ひずみとキャリアドーピングに関する最も近い隣接するMn原子間の距離。 a のバリエーション 角度と c 二軸ひずみに対する距離、 b の変動 角度と d キャリアドーピングに関する距離。キャリアドーピングの正の値と負の値は、それぞれ電子と正孔のドーピングを表します
Mn–MnとMn-Brの両方の距離は、ひずみが–5%から5%に変化するにつれて単調に増加します。これに対応して、図8aに示す二軸ひずみ下の交換パラメータは、二軸ひずみが–5%から5%に変化すると減少し、–5%二軸ひずみ下で最大値(1.18 meV)に達します。 Mn 3 のキュリー温度 Br 8 –5%の二軸ひずみ下の単分子層は160 Kです(図9a)。特に、引張ひずみの増加に伴うMn-Br結合が長くなり、Mn-Br3,4,5,6-Mnの角度が90°から外れることが、FM超交換相互作用が弱くなる主な理由です。その結果、キュリー温度が低下します。 CrPTe 3 と似ています およびFePS 3 単層[79]。さらに、MDEはひずみの増加とともに減少します(追加ファイル1:図S5(b))。 –1%の二軸ひずみ下のMAEが最大です(–3.04 meV)。 –5–5%のひずみは、Mn 3 の大きな構造変形を引き起こしません。 Br 8 単分子層であり、そのバンド構造の形態はほとんど変化しません。 Mn 3 Br 8 単分子層は半金属のままです。半導体スピンチャネル内のVBMとCBMはどちらも、図1および2に示すように、わずかに上向きに移動してより高いエネルギーになります。 8cおよび10;バンドギャップは、5%の二軸ひずみの下で二軸ひずみが3.12eVに増加するにつれてゆっくりと増加します。
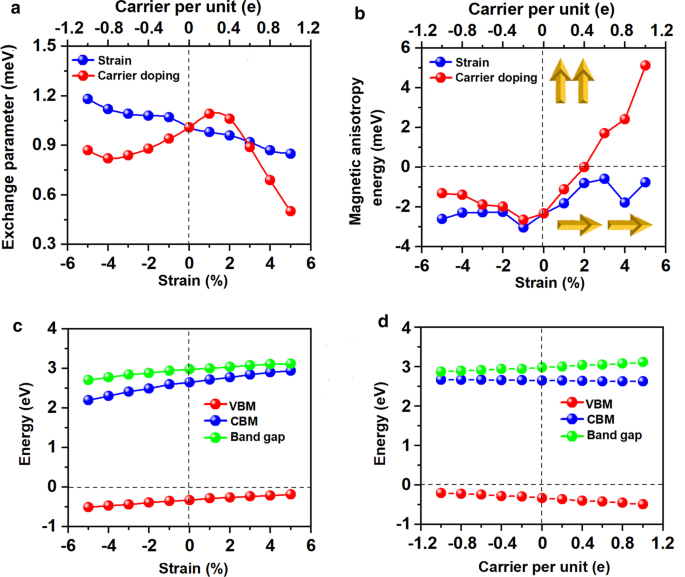
a のバリエーション 交換パラメータと b Mn 3 の磁気異方性エネルギー(MAE) Br 8 適用された二軸ひずみおよびキャリアドーピングに関する単分子層。 Mn 3 の半導体チャネルにおける価電子帯の最大値(VBM)、伝導帯の最小値(CBM)、およびバンドギャップの変動 Br 8 c に関する単分子層 加えられた二軸ひずみと d キャリアドーピングレンジング。キャリアドーピングの正の値と負の値は、それぞれ電子と正孔のドーピングを表します
Mn原子のオンサイト磁気モーメントと比熱 C v Mn 3 のハイゼンベルクモデルに基づく温度の関数として Br 8 単層 a -5%の二軸ひずみの下で、 b 0.2e、 c -0.6e、および d -式単位あたり0.8eのキャリアドーピング。正の値と負の値は、それぞれ電子と正孔のドーピングを表します
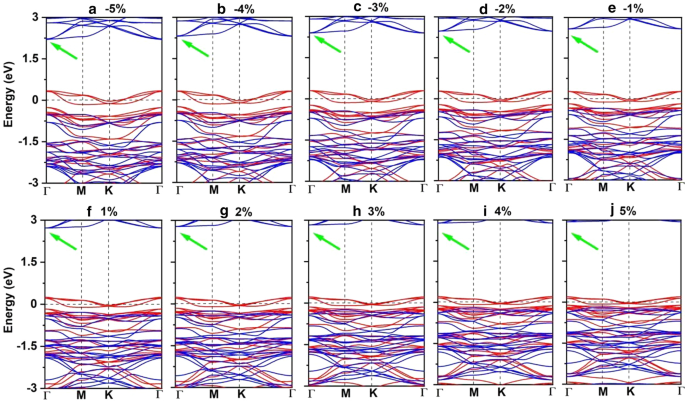
a – j Mn 3 のスピン分解バンド構造 Br 8 -5%から5%の二軸ひずみ下の単分子層。緑の矢印は間接バンドギャップを示します
電子/正孔のドーピングは、常にVBM / CBMがフェルミ準位から離れることにつながります。私たちの計算によると、Mn 3 Br 8 –1–1e(〜\(1.7 \ times 10 ^ {14} {\ text {cm}} ^ {{-2}} \))の単分子層は、式単位あたりのキャリアドーピングがFMのままです。各Mnイオンの原子磁気モーメントはまだ13 /3μ Bです。 図7bおよびdに示すように、式単位あたり–1eから1eのキャリアドーピングでは、Br3,4,5,6原子を含むMn-Br-Mn角度は約90°〜98°です。 Mn-Br1,2-Mnの角度は約88°〜90°です。 Mn–MnおよびMn-Br1,2の距離は、電子ドーピングの増加とともに増加します。 Mn 3 Br 8 0.2eおよび0.4eのキャリアドーピングを備えた単分子層は、より大きな磁気交換パラメータを持っています(図8a)。 0.2e電子ドーピングでのキュリー温度は140Kの中で最大です(図9b)。さらに、–1e〜0.2eドーピングでは、MAEは面内方向に沿っています。 MDEは、電子ドーピングの増加とともに減少します。 0.4eドーピングでは、MCEは正になり、式単位あたり0.41meVの値になります。 MDEを考慮した場合、MAEは式単位あたりわずか0.01 meVです(追加ファイル1:図S5(a)および(b))。 0.6e、0.8e、および1eのドーピングでは、PMA(垂直磁気異方性エネルギー)はそれぞれ1.70、2.42、および5.13 meVであり、スピントロニクスアプリケーションに十分な大きさです(図8b)。
さらに、Mn 3 Br 8 式単位あたり–1e〜1eのキャリアドーピングを備えた単分子層は、ハーフメタリックのままです。図8dに示すように、半導体スピンチャネルのバンドギャップは、電子/正孔のドーピングの増加に伴ってわずかに増加/減少します。 VBMとCBMの位置は変わりません。例外的、Mn 3 Br 8 単分子層はFMスピンギャップレス半導体(SGS)になり、金属スピンチャネルは–0.6eおよび–0.8eの正孔ドーピングの下で非常に小さなエネルギーギャップ(0.07 eV)を開きます。そのフェルミ準位はバンドギャップ領域にあります(図11bおよびc、より明確な図は追加ファイル1:図S6(a)および(b)に示されています)。これに対応して、電子は小さなエネルギー入力で価電子帯から伝導帯に容易に励起され、100%スピン偏極した電子と正孔のキャリアを同時に生成します。 –0.6eおよび–0.8eの正孔ドーピングでのキュリー温度は110 Kであり(図9cおよびd)、液体窒素温度(77 K)よりも高くなっています。 \(10 ^ {13} \ sim10 ^ {15} {\ text {cm}} ^ {-2} \)の電荷密度変調がすでに実験的に達成されていることを考慮すると[80,81,82]、予測される特性Mn 3 の Br 8 キャリアドーピングを伴う単分子層も実験的にアプローチ可能です。
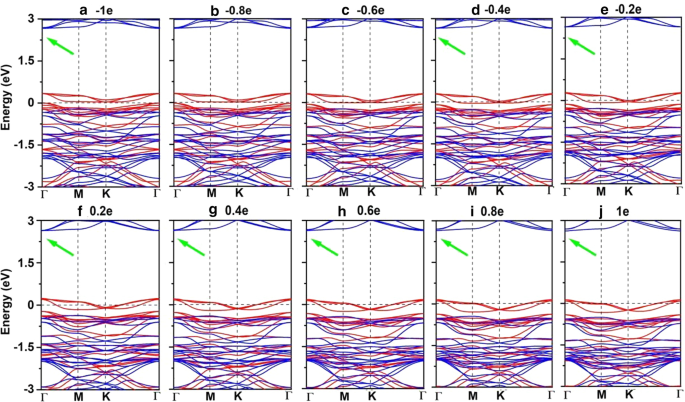
a – j Mn 3 のスピン分解バンド構造 Br 8 式単位あたり-1eから1eまでのキャリアドーピングを伴う単分子層。正の値と負の値は、それぞれ電子と正孔のドーピングを表します。緑の矢印は間接バンドギャップを示します
結論
要約すると、Mn 3 の安定性、電子的、および磁気的特性 Br 8 単層は注意深く調査されています。私たちの結果は、Mn 3 Br 8 単分子層はFMハーフメタルで、キュリー温度は130 K、半導体スピンチャネルのバンドギャップは2.97eVです。さらに、各Mnイオンの磁気モーメントは13 /3μ B です。; MAEは、式単位あたり–2.33meVです。 Mn 3 Br 8 単分子層は、MnBr 2 の\({2} \ times {2} \ times {1} \)スーパーセルに単一のMn空孔を誘導することによって設計されています。 AFM結合を破壊する単分子層d 5 構成。 Mn空孔を形成する可能性とMn 3 の動的で機械的な安定性 Br 8 単層が包括的に確認されています。さらに、Mn 3 Br 8 二軸ひずみ下の単分子層–5%〜5%は依然としてFMハーフメタルであり、半導体スピンチャネルのバンドギャップは2.71〜3.12 eVであり、–5%二軸ひずみ下のキュリー温度は160Kです。二軸ひずみとキャリアドーピングの両方により、 MAEが増加し、電子ドーピング下で平面に垂直になります。 0.8eおよび0.6eの正孔ドーピングでは、Mn 3 Br 8 単分子層は、バンドギャップが0.07 eVのスピンギャップレス半導体(SGS)になります。私たちの計算は、Mn 3 を示しています Br 8 キュリー温度が高く、MAEが大きく、磁気モーメントが大きく、2軸ひずみとキャリアドーピングが適用された状態で電子的および磁気的特性を調整できるFMハーフメタルとしての単分子層。
データと資料の可用性
現在の研究中に生成および/または分析されたデータセットは、合理的な要求に応じて対応する著者から入手できます。
略語
- 2D:
-
二次元
- AFM:
-
反強磁性
- CBM:
-
伝導帯の最小値
- DFT:
-
密度汎関数理論
- DOS:
-
状態密度
- FIM:
-
フェリ磁性
- FM:
-
強磁性
- GGA:
-
一般化された勾配近似
- GKA:
-
Goodenough–Kanamori–Anderson
- MAE:
-
磁気異方性エネルギー
- MCE:
-
磁気結晶異方性エネルギー
- MC:
-
モンテカルロ
- MDE:
-
磁気双極子異方性エネルギー
- MTJ:
-
磁気トンネリングジャンクション
- NM:
-
非磁性
- NN:
-
最寄りの
- PAW:
-
プロジェクター拡張波
- PBE:
-
Perdew–Burke–Ernzerhof
- PMA:
-
垂直磁気異方性エネルギー
- PNGN:
-
多孔質窒素ドープグラフェンネットワーク
- SGS:
-
スピンギャップのない半導体
- SOC:
-
スピン軌道相互作用
- VASP:
-
ウィーンab-initioシミュレーションパッケージ
- VBM:
-
価電子帯の最大値
- VDW:
-
ファンデルワールス
ナノマテリアル
- PersuasionIncのCraigTrevorへのインタビュー。
- パイロットから大規模な展開へ:IoTで距離を置く
- 弱い強磁性配向を伴う反強磁性秩序を有するNiドープSb2Te3トポロジカル絶縁体の磁化率分岐
- 半極性InxGa1-xN / GaN多重量子井戸を備えた紫外線GaNベースのフォトニック準結晶ナノピラミッド構造からの多色発光
- 単層MoS2と六角形窒化チタンナノディスクアレイを備えたブロードバンドパーフェクトアブソーバー
- 水からのスルホンアミド除去のための再利用可能な吸着剤としての磁性炭素ミクロスフェア
- 非対称コンタクトフォームを備えた多層MoTe2フォトトランジスタからの顕著な光起電力応答
- 粗い表面の通常の荷重による接触面積の変化:原子スケールから巨視的スケールまで
- メタマテリアルの複数の磁気双極子共鳴からの光周波数での単層グラフェンのマルチバンドおよびブロードバンド吸収増強
- 全炭素3Dナノ濾過膜による水からの抗生物質の除去
- 熱制御されたRKKY相互作用によるナノ構造の磁気ヒステリシス