


リチウムイオン電池用の高性能アノード材料としてのペンタシリグラフェンの第一原理研究
要約
第一原理計算から、新しい五角形のSi / Cの複雑さは、リチウムイオン電池の有望なアノード材料としての潜在的な用途があると予測されています。ペンタシリグラフェン(P-Si 2 C 4 )は、C原子のみで構成されるペンタグラフェンよりも優れています。電子バンド構造分析は、空のC-2 p z P-Si 2 の状態 C 4 Liからの電子を収容および安定化するためのスペースを提供します。これにより、Liの貯蔵がエネルギー的に有利になります。その結果、P-Si 2 の1つの式単位で4つのLi原子を格納できます。 C 4 、1028.7 mAhg -1 の理論的な重量分析Liストレージ容量に対応 。 Li吸着P-Li x の金属電子構造 Si 2 C 4 非常に小さいLi移行エネルギー障壁は、バッテリーの急速充電/放電性能に有益です。 P-Si 2 へのLi吸着相互作用のメカニズム C 4 議論されています。これらの結果は、高性能リチウムイオン電池用の2次元Si / C複合アノード材料を設計するための新しい戦略を示しています。
背景
現在市販されているリチウムイオン電池(LIB)の比較的低いエネルギー密度は、商用電気自動車(EV)の要件を満たすことが難しく、EV産業の発展にとって大きな課題となっています[1、2]。 LIBのエネルギー密度を高めるには、電極材料の容量を改善する必要があります。グラファイトは非常に優れたサイクリング性能を備えているため、最も広く使用されているアノード材料ですが、理論上の重量分析能力(372 mAhg -1 )は比較的低い[3、4]。一方、シリコンの理論重量分析能力は約4200 mAhg -1 と非常に高くなっています。 [5]ですが、完全にリチウム化された状態で最大420%の非常に大きな体積膨張があるため、サイクリング性能は低くなります[6]。シリコンアノードとカーボンアノードの両方を活用するには、Si / C複合アノードの設計は、学術的にも技術的にも重要です。
C原子が部分的にSi原子に置き換えられた2次元(2D)グラフェンのような層状材料であるシリグラフェンは、第一原理計算から安定した2D材料であると最初に予測され[7,8,9,10]、実験から首尾よく準備された[11、12]。リンら。は、2DSiCシートが溶液剥離技術によって調製できることを示しました[11]。彼らはまた、準2D SiC 2 の準備にも成功しました。 空気中で数ヶ月間保存できるシート[12]。後で、第一原理計算は、シリグラフェンが1520 mAhg -1 の理論容量を提供する有望なアノード材料であることを示唆しています。 および1286mAhg -1 g-SiC 5 の場合 およびg-SiC 2 、それぞれ[13]。シリグラフェンアノードは、グラファイトアノードからの高いサイクル安定性と、シリコンアノードからの高容量を継承していることが示されています。予測される高いLi貯蔵容量は、 sp からのSi原子の変化に関連する、シリグラフェン単分子層とのLi吸着相互作用の強化に起因します。 2 sp へ 3 -[13]のように。ただし、電子配置は sp から変更されます 2 sp へ 3 -likeは、シリグラフェンへのLi吸着中に明らかな構造変化を伴います。これは、LIBのアノード材料としてのシリグラフェンのサイクル性能には適していません。より良い解決策は、すでに sp を持っているSi / C複合材料を設計することです。 3 -電子配置のように。
炭素には多くの種類の同素体があり、 sp で形成されます 、 sp 2 、および sp 3 ハイブリダイゼーションまたはそれらの組み合わせ。非常に安定した sp 2 加えて、グラファイトカーボンの大きなπ結合電子配置は、グラフェンへの弱いLi吸着相互作用の原因です。単層グラフェンにLiが吸着すると、Liからグラフェン層への電荷移動が発生します[14]。次に、Liは正に帯電し、魅力的なクーロン相互作用によってグラフェン層に結合します。ただし、グラフェン層のLiからの過剰な電荷は、グラフェンの大きなπ結合を破壊します。これは、エネルギー的に不利です。その結果、単層グラフェンへのLi吸着は、体心面の凝集エネルギー( bcc )よりも負に高い吸着エネルギーでは有利になりません。 )リチウムイオン電池では許可されていない相リチウム金属。その結果、元のグラフェン単分子層によるLiの貯蔵は許可されていません[15]。あるいは、ハードカーボン材料は、グラファイトカーボン材料と比較してはるかに高いLi / Na貯蔵重量分析能力を提供します[16、17、18]。ハードカーボン材料は、両方の sp で構成されるアモルファス相として知られています。 2 および sp 3 炭素原子[19]。ハードカーボン材料のより高いLi貯蔵重量分析能力が sp に関連している可能性はありますか? 3 電子構成?
sp として知られるペンタグラフェン 2 - sp 3 ハイブリッド2D炭素同素体[20]は、第一原理計算[21]から、Li / Naイオン電池の有望なアノード材料であると予測されました。 2D炭素同素体として、ペンタグラフェンは、ハニカム構造の従来のグラフェンと比較して、はるかに強いLi吸着挙動を示します。この異なるLi吸着挙動は、 sp にも関連していますか? 3 -ペンタグラフェンの電子配置のように?答えが「はい」の場合、その背後にある固有のメカニズムは何ですか?
ペンタグラフェンは動的に安定した炭素同素体であると予測されていますが、その凝集エネルギーは、世界で最も安定な相(グラファイトまたはグラフェン)と比較して大幅に高くなっています。ペンタグラフェンの凝集エネルギーは、単層の六角形グラフェンよりも原子あたり約0.9 eV高く[20、22]、工業的にペンタグラフェンを大規模に製造することは(可能であれば)非常に困難です。しかし、アノード材料としての用途に関しては、大規模な製造が非常に重要です。座屈はシリセンに見られるため、Siは sp でより安定していることに注意してください。 3 sp よりもハイブリダイゼーションのように 2 [23,24,25]一方、C原子は sp を好みます 2 2D構造でのハイブリダイゼーション; sp を置き換えると推測するのは合理的です 3 ペンタグラフェンの構造にSi原子を含むC原子のようにエネルギー的に支持されます。この構造をペンタシリグラフェンと呼びます。最近の実験では、五角形のSiベースのナノリボンがAg上で成長できることが示され(110)[26]、Siベースの五角形構造の形成が実験的に可能であることを示しています。
理論的には、ペンタシリグラフェン(P-SiC 2 )の電子的および結合的性質 )はLopez-Bezanillaらによって研究されました。そして彼らは、P-SiC 2 p-p-σおよびp-p-π電子バンドの垂直順序の部分的な反転を示します[27]。その後、P-SiC 2 の電子輸送特性 研究され、ペンタグラフェンおよびペンタCN 2 と比較されます [28]。興味深いことに、P-SiC 2 の電子輸送性能が実証されています ひずみ工学によって調整することができ、一軸圧縮ひずみは単分子層ペンタ-SiC 2 の正孔移動度を高めることができると予測されました。 最大1.14×10 6 cm 2 V -1 s -1 [29]。構造の類似性にもかかわらず、ペンタシリグラフェンはペンタグラフェンと比較して異なる輸送特性を持っています。それはHuらによって発見されました。ペンタグラフェンの熱伝導率は、伸縮によって標準的な単調な還元を示しますが、ペンタ-SiC 2 異常な非単調な上下動作を持っています[30]。ペンタシリグラフェンのこれらの興味深い特性は、構造内のSi原子の電子的および化学的性質に強く関連しています。シリセンへのLi吸着相互作用はグラフェンへの相互作用に比べてはるかに強いため、Si元素自体がLi吸着を強化するのに有益であることがわかりました[31、32]。したがって、ペンタシリグラフェンをLIBのアノード材料として使用できるかどうかを知ることは興味深いかもしれません。
この研究では、第一原理計算を用いてペンタシリグラフェンのLiイオン貯蔵挙動を調査し、ペンタシリグラフェンがLiイオンを貯蔵するメカニズムについて具体的に説明します。ペンタシリグラフェンの熱力学的安定性から研究を開始し、続いてその上へのLi吸着の固有の相互作用の詳細な分析を行います。最後に、LIBのアノード材料としてのペンタシリグラフェンの性能について説明します。
計算方法
この作業のすべての計算は、密度汎関数理論(DFT)に基づくVienna Ab initio Simulation Package(VASP)[33]を使用して実行されます。 Perdew-Burke-Ernzerhof(PBE)によってパラメーター化された一般勾配近似(GGA)交換および相関汎関数と組み合わせたプロジェクター拡張波(PAW)法[34、35]が使用されます[36]。平面波のカットオフエネルギーは、すべての計算で450eVになるように選択されています。格子定数とイオン位置は完全に緩和され、最終的な力は0.02 eV /Åに収束します。電子バンド構造は、Heyd-Scuseria-Erznerhof(HSE06)混成汎関数[37]を使用して計算されます。これは、混成汎関数が電子構造をより正確に記述しているためです。状態密度(DOS)の計算は、0.05eVのスミアリング幅でガウススミアリング法によってスミアリングされます。モンホルストパック[38] k -ポイントサンプリングが使用され、 k の密度 -メッシュは0.05Å -1 よりも厚い 非経験的分子動力学(AIMD)シミュレーションおよび0.03Å -1 の場合 他の計算のため。原子電荷分布は、Bader電荷分析[39]で分析されます。リチウムイオン移動経路は、クライミングイメージナッジドエラスティックバンド(CINEB)法[40]で最適化されています。吸着エネルギー E ad 計算方法:
$$ {E} _ {\ mathrm {ad}} =\ left({E} _ {\ mathrm {host} + n \ mathrm {Li}}-{E} _ {\ mathrm {host}}-{nE } _ {\ mathrm {Li}} \ right)/ n $$ここで E ホスト 、 E Li 、および E host + Li は、それぞれホストのペンタシリグラフェン材料、Li原子、およびLi吸着ホストの総エネルギーです。 n ペンタシリグラフェンに吸着されたLiイオンの数を示します。ファンデルワールス(vdW)相互作用の吸着エネルギーへの影響は、Becke-Jonson減衰を使用したDFT-D3法を使用してテストされます[41]。吸着エネルギーに加えて、平均Liインターカレーションポテンシャル(vs Li + / Li)は、 V からのLi金属(bcc相)の吸着エネルギーと凝集エネルギーの差から直接得ることができます。 ave =−( E ad − E Li −凝集性 )、エネルギーとポテンシャルの単位としてそれぞれeVとVを選択した場合。
結果と考察
Penta-siligrapheneの構造と安定性
ペンタグラフェンの構造(図1aを参照、P-C 6 として示される) この論文の以下で)はP-42 1 を所有しています m 対称性(空間群No.113)。最適化された格子定数は a = b =3.636Å、以前の結果と一致[20、21]。この構造には、4配位炭素(図1aではC1と表示)と3配位炭素(図1aではC2と表示)の2種類の炭素原子があります。炭素原子の局所的な形状から、C1が sp であることがわかります。 3 -C2が sp である間に混成のように 2 -混成のように。 C2アトムは sp と見なされますが 2 混成軌道[20]のように、二重のC2–C2結合の特徴により、C2原子の化学的特性がグラフェンの化学的特性とは異なります。これについては、この論文の次の記事で詳しく説明します。 P-C 6 のC1原子をSi原子に置き換える 構造では、ペンタシリグラフェンが形成され(図1bを参照、最適化された構造の結晶情報ファイルは追加ファイル1:補足資料のSI-1に記載されています)、P-Si 2 > C 4 この論文の次の部分で。 Si原子の原子半径はC原子の原子半径よりも大きいため、P-Si 2 の格子定数 C 4 ( a = b =4.405Å)はP-C 6 よりも大きい 、他の報告された結果とよく一致している間[27,28,29,30]。
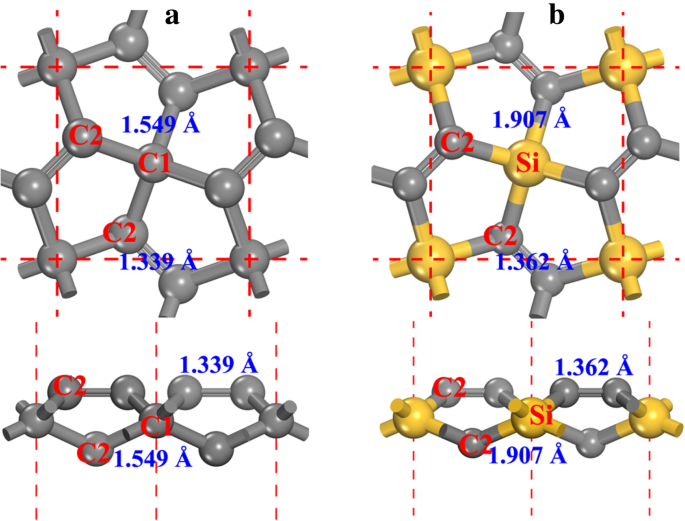
a ペンタグラフェンと b の球棒モデル ペンタシリグラフェン。上面図(上)と側面図(下)の両方が表示されます。灰色の球と黄色の球は、それぞれC原子とSi原子です。 4配位および3配位の炭素原子は、それぞれC1およびC2として示されます。結合長も各結合と一緒に表示されます
相対的な熱力学的安定性を評価するために、表1に、C、Si、およびC / Si錯体のさまざまな同素体の凝集エネルギーを示します。ペンタグラフェン(P-C 6 )は、ab initio分子動力学(AIMD)シミュレーション[20]、P-C 6 の凝集エネルギーから、1000Kで安定していることが示されています。 (− 8.24 eV・atom -1 )は単層グラフェンに比べてはるかに高い(− 9.14 eV・atom -1 )。これは、P-C 6 の大量生産を示しています。 非常に難しいに違いありません。一方、P-Si 2 の凝集エネルギー C 4 (− 7.26 eV・atom -1 )は、最も安定した同素体g-Si 2 と比較してわずか0.2eV高いです。 C 4 (− 7.46 eV・atom -1 )、ペンタシリグラフェンの調製はP-C 6 と比較してはるかに簡単である可能性があることを示しています 。 P-Si 2 の構造安定性を検証するには C 4 、P-Si 2 のフォノン分散曲線 C 4 計算され、図2に示されています。ただし、Γ点(0.0039THzまたは0.13cm -1 )の近くの小さな領域に小さな虚数周波数が見られます。 )、これらの小さな虚数周波数(1 cm -1 以下)が一般的に受け入れられているため、システムは動的に安定していると信じることができます。 )はシミュレーションのアーティファクトである可能性があります[42]。架空の周波数は、ゲルマネン[43]やアルセネン[44]などの他の動的に安定した2D材料でも報告されています。計算の精度を上げる、または別の計算方法を使用するなどの技術処理を適用すると、これらの虚数周波数を削除できます。
<図>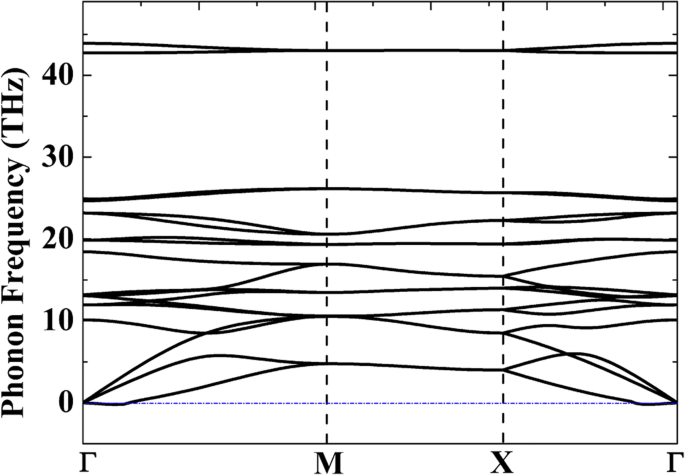
2D P-Si 2 のフォノン分散曲線 C 4 線形応答理論から計算された単分子層
さらに、AIMDシミュレーションも実行され、P-Si 2 の構造安定性を評価します。 C 4 高温で。 AIMDは、1000 K、1500 K、2000 K、および2500 Kの温度で標準アンサンブル内の3×3および4×4スーパーセルを使用して実行されます(追加ファイル1:図S1を参照)。追加ファイル1:図S2およびS3は、P-Si 2 の原子構成を示しています。 C 4 それぞれ3×3および4×4スーパーセルを使用したさまざまな温度でのAIMDシミュレーションの最後に。示されているように、20psのシミュレーション時間中に温度が2000Kに達すると、五角形の原子環は変化せず、構造が2000 Kに耐えることができることを示しています。一方、深刻な構造変形が観察され、六角形のリング(追加ファイル1:図S2dを参照)およびその他の欠陥(追加ファイル1:図S3d)がスナップショットに表示され、構造が2500Kで破壊されたことを示しています。P-Siで見つかった六角形のリング 2 C 4 2500 Kで、g-Si 2 C 4 (六角形のリングで構成されている[13])は、五相P-Si 2 よりも安定しています。 C 4 、表1に示されている凝集エネルギーと一致します。これらの結果は、P-Si 2 の構造安定性を確認しています。 C 4 P-C 6 に比べてはるかに安定しています 、1000Kの温度にしか耐えられません。
Penta-siligrapheneへのLi吸着
ペンタシリグラフェンP-Si 2 へのLi吸着を研究するために C 4 、異なるLi吸着サイトが考慮され、緩和後に4つの安定した吸着サイト(図3aに示す)が見つかります。安定したLi吸着サイトは、Si原子のトップサイト(Tと表示)、Si 2 の中空サイト(Hと表示)です。 C 3 五角形のリング、および下層(B1)と上層(B2)の2つのC2原子間のブリッジサイト。これらのサイトでのLiイオン吸着の優先度は、表2に示す吸着エネルギーによって特徴付けることができます。結果は、最も安定したLi吸着サイトがB1サイトであり、吸着エネルギーが-1.922eVであることを示しています。一方、Hサイトでの吸着エネルギー(-1.905 eV)はB1サイトに非常に近いです。吸着エネルギーが低いほど吸着高さが低くなるため、Li吸着エネルギーも吸着高さで表されます(表2を参照)。 Li吸着プロセスの開始時には、Liイオンが最も安定したB1サイトに留まることが好ましい。すべてのB1サイトが占有された後(Li 2 の化学量論に対応) Si 2 C 4 図3b)を参照すると、LiイオンはHサイトに留まり始めます。 B1サイトとHサイト間の距離が非常に小さいため(〜1.5Å)、B1サイトとHサイトのLiイオンに対して強い反発相互作用が発生します。その結果、B1サイトのLiイオンは近くのHサイトに反発するため、すべてのHサイトがLi 4 の状態で占有されている間、B1サイトは空になります。 Si 2 C 4 (図3cを参照)。 Li吸着エネルギーへのvdW相互作用の影響もテストされ、結果は表2の括弧内に示されています。示されているように、vdW相互作用はさまざまな吸着サイトの吸着エネルギーに-0.12から-0.17eVに寄与します。 vdW相互作用がLi吸着に有利であることを示しています。
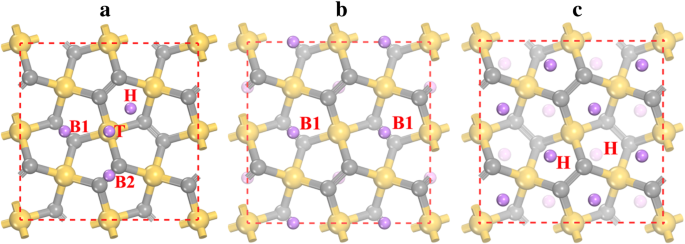
a のLi吸着サイト ペンタシリグラフェン表面と b の最も安定した構造の原子配置 Li 2 Si 2 C 4 および c Li 4 Si 2 C 4 。黄色(最大)、灰色(中サイズ)、および紫色(最小)の球は、それぞれSi、C、およびLi原子です。 H、T、B1、およびB2はLi吸着サイトを示します
Li吸着エネルギーと bcc の凝集エネルギーの比較 相Li金属(− 1.86 eV・atom -1 )、Li吸着が電気化学的に活性であるかどうかを判断することができます。吸着エネルギーがLi金属の凝集エネルギーよりも低い場合、Liイオン吸着が優先され、吸着は正の放電電位に対応します。表1に示すように、B1サイトとHサイトの両方の吸着エネルギーは-1.86 eV未満であり、B1サイトとHサイトの両方がLi貯蔵の電気化学的に活性なサイトであることを示しています。 Liの貯蔵容量を評価するために、さまざまなLiイオン濃度でのLi吸着エネルギー x (Li /(Si + C)比)が計算され、 bcc の凝集エネルギーと比較されます。 李金属。図4に示すように、Li / C比 x の場合、Li吸着エネルギーは-1.86eVより低くなります。 P-Si 2 の1つのユニットセルに吸着された4つのLi原子に対応する2/3よりも小さい C 4 理論重量分析容量は1028.7mAhg -1 (Li 4 Si 2 C 4 )。容量に出力電圧を掛けたものに等しいバッテリーのエネルギー密度は、Liストレージ容量と比較してより懸念されます。優れたアノード材料は、吸着エネルギーから得られる比較的低い電気化学ポテンシャルを持つ必要があります。平均電位は約0.1〜0.2 Vであり、これは比較的低く、フルバッテリーシステムのより高い出力電圧に有益です。さらに、図4は、さまざまなLi吸着濃度での格子定数の変化も示しています。示されているように、P-Si 2 の格子定数 C 4 Li吸着時にわずかに収縮します。濃度が x の場合 が1/6の場合、格子変化は最大値に達し、-0.94%と小さくなります。これは、充電/放電プロセス中の体積変化が非常に小さいことを示しています。わずかな体積変化は、P-Si 2 の構造を維持するのに役立ちます。 C 4 サイクリング中に安定するため。
P-Si 2 で計算されたLi吸着エネルギー C 4 Li吸着濃度(Li /(C + Si)比)の関数としての表面(赤いサイクル、左側の軸)と格子定数の変化(青い四角、右側の軸)。 bcc の凝集エネルギー 比較のためにリチウム金属相も含まれています
Li吸着時のペンタシリグラフェンの電子構造分析
ペンタシリグラフェン(P-Si 2 )の電子バンド構造 C 4 )とそのリチウム化状態を図5に示します。ご覧のとおり、P-Si 2 C 4 は、約2.35 eVの間接バンドギャップを持つ半導体であり、ペンタグラフェンP-C 6 の3.46eVと比較してはるかに小さいです。 (追加ファイル1:図S4を参照)。 P-Si 2 のバンドギャップが小さい C 4 P-C 6 より これは、特に対称性の高いM点とΓ点で、最も高い占有帯域(図5aの11番と12番)の分散が強化されていることに起因しています。バンドギャップは、No.12(最も高い占有バンド)とNo.13(最も低い非占有バンド)のエネルギーバンドの間に開かれます。バンドNo.12のエネルギー準位はM点で大幅に上昇し、フェルミ準位が上昇し、バンドギャップが減少します。
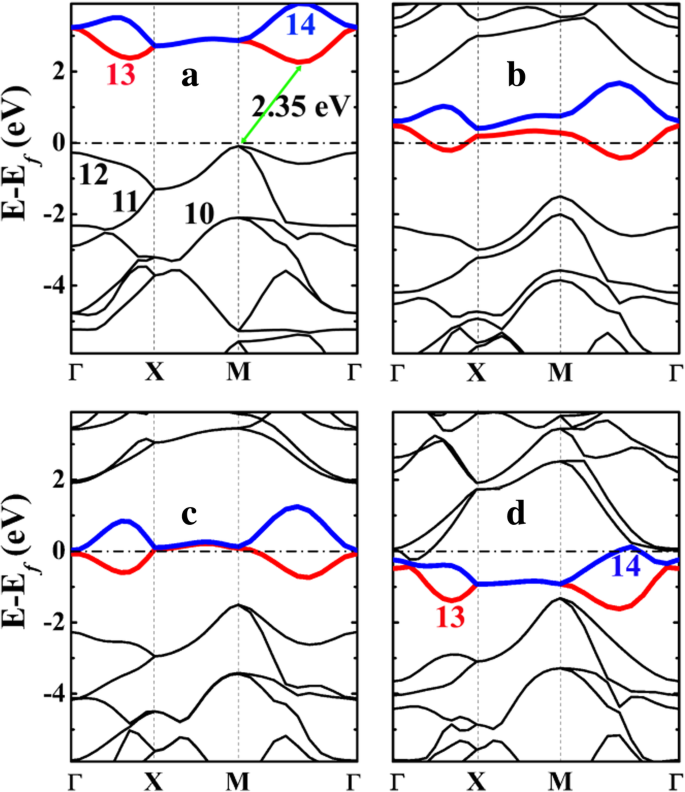
a の電子バンド構造 ペンタシリグラフェンP-Si 2 C 4 とそのリチウム化された状態 b P-LiSi 2 C 4 、 c P-Li 2 Si 2 C 4 、および d P-Li 4 Si 2 C 4 HSE06から計算されます。フェルミ準位は0eVになるように選択されています。 a の10から14までの数字 および d バンド番号を示し、バンド番号13と14はそれぞれ赤と青で強調表示されます。バンドのラベリングはVASPコードに準拠しており、バンドのラベリングは価電子帯と伝導帯を参照しており、コア電子はラベリングに含まれていません
。図6に示すバンドNo.10〜14に投影された電荷密度(波動関数)の形状を分析すると、バンドNo.12がCとSiの間に形成されたσ結合の結合状態に対応していることがわかります。一方、バンドNo. 13(および14)は2- p に対応します。 z C原子の状態。空のC- p z 状態は、Li吸着からの電子を収容および安定化するためのスペースを提供し、Li吸着プロセスをエネルギー的に有利にします。
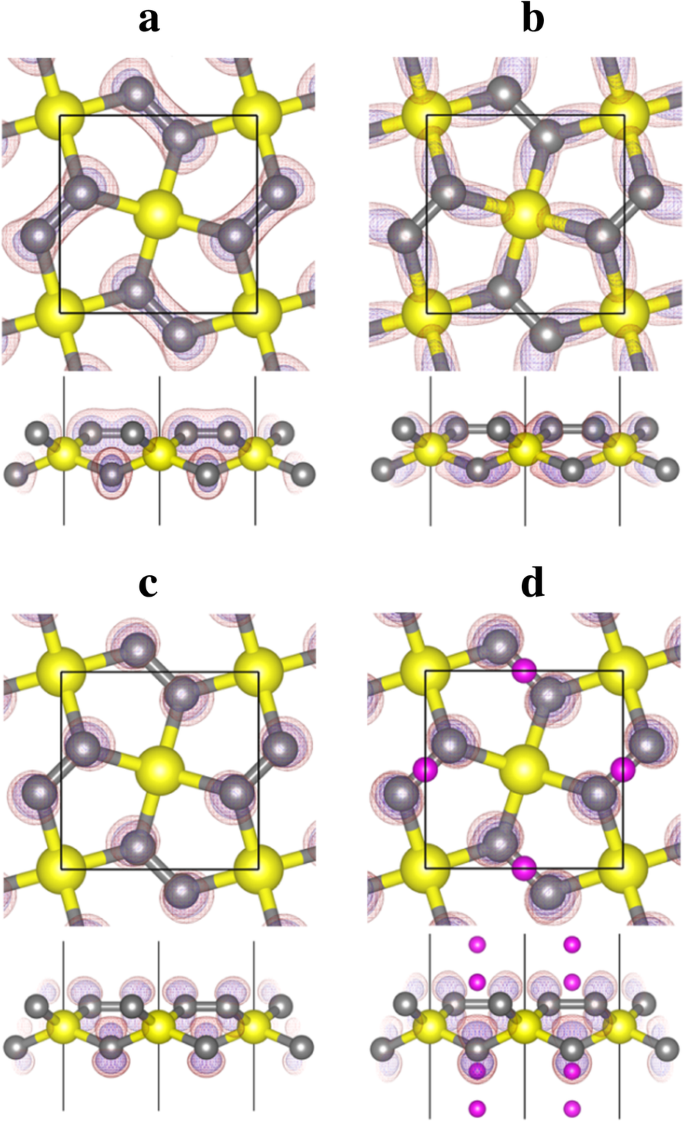
バンド a のバンド分解電荷密度コンター 10番、 b No. 12、および c ペンタシリグラフェンの13番(P-Si 2 C 4 、図5a)およびバンド d リチウム化ペンタシリグラフェン(Li 4 Si 2 C 4 、図5d)。黄色(大)、灰色(中型)、紫色(小)の球は、それぞれSi、C、Li原子です。電荷密度の等高線は透明な赤で表示されます(等値面値は0.02 e /Å 3 )および青(等値面値0.01 e /Å 3 )色
P-Si 2 の最高占有エネルギーバンドの分散の強化 C 4 2つの要因に起因する可能性があります。1つは、炭素のみのペンタグラフェンPC 6と比較して、バンドNo. 12(C–Siσ結合)と正に帯電したSi原子を占める電子間のクーロン引力です。 。より悪い電荷分析は、Si原子がペンタシリグラフェンP-Si 2 で正に帯電していることを示しています C 4 。表3に示すように、シリグラフェンまたはペンタシリグラフェンのいずれかのSi原子のバダー電荷は約1.65 e です。 、Si原子が+ 2.35 e で正に帯電していることを示しています 。一方、ペンタグラフェンのC1原子(P-C 6 )も正に帯電していますが、正味の電荷は+ 0.08 e のみです。 。したがって、共有結合相互作用に加えて、C原子とSi原子の間の強いクーロン相互作用がP-Si 2 で発生します。 C 4 、P-C 6 との比較 。これは、フェルミ準位近くの占有エネルギーバンド(No. 12)の分散に有益です。第二に、P-Si 2 の座屈の強化 C 4 座屈が大きいほど sp が大きくなるため、バンドNo. 12(C–Siσ結合の結合状態)の分散にも寄与する可能性があります。 3 -混成でより強いσ結合がC–Si原子間に形成されます。これは、P-Si 2 の安定した構造安定性の重要な理由でもあります。 C 4 P-C 6 との比較 。さらに重要なことに、Si原子とC原子の間の座屈は、Liの吸着とともに増加し、リチウム化状態のP-Li 4 で0.876Åになります。 Si 2 C 4 。この状態では、SiC 4 のC–Si–C(102.50°および124.59°)結合角 四面体は標準の四面体の108.47°に近づき、 sp 3 -Liの吸着により、Si原子の混成軌道とC–Si結合の強度が強くなります。その結果、図4に示すように、バンドNo. 12の分散も、Li吸着濃度の増加に伴って向上します。
<図>ペンタシリグラフェン(P-Si 2 )の表面にLiが吸着すると C 4 )、Li原子からの電荷移動(実際のバッテリー動作では、Li + イオンは内部回路から発生し、同じ量の電子は外部回路から発生します)P-Si 2 の炭素原子 C 4 。その結果、図6b–dに示すように、過剰な電子が占有されていないバンド(バンドNo. 13および14)を下って移動し、システムの金属電子構造を生じます。金属製の電子構造により、P-Si 2 の良好な電子伝導性が保証されます。 C 4 充電/放電プロセス中のアノード。これは、P-Si 2 を使用するバッテリーシステムのレート性能に有益です。 C 4 アノード。
上記で説明し、図6のバンド投影電荷密度で示されているように、バンドNo.13と14は p です。 z P-Si 2 のC原子の状態 C 4 。これらの空のバンドは、Li吸着にとって非常に重要です。正に帯電したLiイオンと負に帯電したP-Si 2 の間のクーロン引力に加えて C 4 基板、C- p を占める電子 z 状態は、正に帯電したSi原子に対して強いクーロン引力を持っています(これにより、バンドNo. 13および14のエネルギーレベルが下がり、基板の総エネルギーが低下します)。結果として、P-Si 2 へのLiの吸着 C 4 エネルギー的に有利です。 P-Si 2 のユニットセルとして C 4 4個のC原子を含み、4個のLi原子がP-Si 2 に吸着できると予想されます。 C 4 水面。 C- p の後 z 状態は完全に占有され、P-Si 2 へのより多くのLi原子の吸着 C 4 精力的に不利になります。これは、図4に示されている計算された吸着エネルギーと一致しています。
P-Si 2でのリチウムイオン移行ダイナミクス C 4
P-Si 2 のレートパフォーマンス C 4 アノードは、電子伝導とリチウムイオン拡散のダイナミクスによって決定されます。上記のように、元のP-Si 2 の電子構造は C 4 は絶縁体であり、Li濃度が低くてもLi吸着時に自然に金属になります。したがって、電子伝導率は、アノード材料としての用途に十分なものでなければなりません。次に、ペンタシリグラフェン上のLiイオン拡散が速度制御ステップになります。 P-Si 2 の構造として C 6 ペンタグラフェン(P-C 6 )と同様です。 )Liイオンが非常に速く拡散する[21]場合、P-Si 2 でのLiイオン拡散が予想されます。 C 6 非常に高速になることもあります。
バッテリーのレート性能は、充電状態(SOC)に強く関係しています。つまり、Liイオンの拡散はLi吸着濃度に依存します。異なるSOCでのLiイオン拡散ダイナミクスを評価するために、2つの極端な濃度、つまり希薄Liイオンと希薄Li空孔が考慮されます。希薄なLiイオン拡散のシミュレーションでは、1つのLiイオンがP-Si 2 のスーパーセルに吸着されます。 C 4 。 「ペンタシリグラフェンへのLi吸着」のセクションでの上記の説明から、図7に示すように、LiイオンはLi濃度が低いときにB1サイトに留まるのを好みます。構造の対称性を考慮すると、Li移動経路は1つだけです(図7)でPath-1として示されているものを見つけることができ、Path-1はP-Si 2 上に完全な2DLi拡散ネットワークを形成します。 C 4 水面。 P-Si 2 上の希薄なLiイオン移動エネルギー障壁 C 4 NEB法に沿って最適化された経路は約0.117eVであり、P-C 6 の経路よりも小さくなっています。 (0.17 eV、Path-II)[21]およびg-Si 2 C 4 (0.548 eV)[13]。 SOCが50%になると、つまり、材料はLi 2 の状態に排出されます。 Si 2 C 4 、すべてのLiイオンは依然としてB1サイトを占有しているため(図3bを参照)、Liイオンの拡散経路は希薄なLiイオンの場合と同じです。これらの結果は、放電プロセスの最初の半分でリチウムイオンの拡散が非常に速くなる可能性があることを示しています。
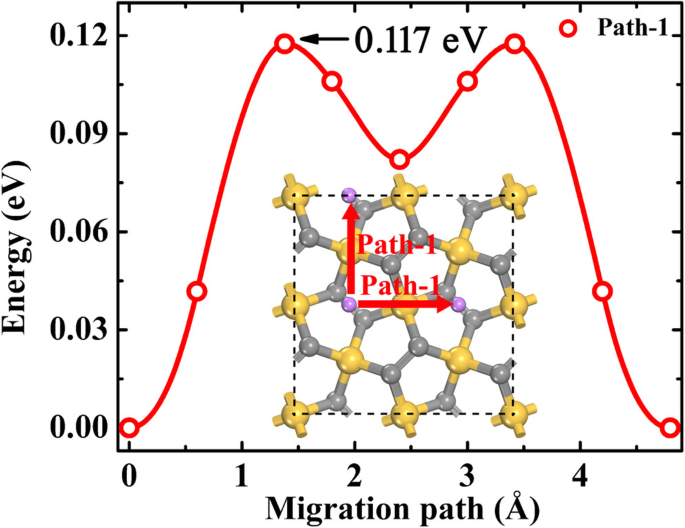
希薄なリチウムイオン移動経路と、P-Si 2 の経路に沿った対応するエネルギープロファイル C 4 。灰色(中型)、金色、紫色の球は、それぞれC、Si、Li原子です。赤い矢印は、2次元拡散ネットワークを示しています
希薄なLi空孔の場合、完全にリチウム化された状態のLi 4 から1つのLiイオンを除去します。 Si 2 C 4 スーパーセルに1つの希薄なLi空孔を作成します。上記のように、Liイオンは、Li濃度が高い場合にHサイトを占有することを好みます(図3cを参照)。したがって、図8aに示すように、3つの異なるLi空孔移動経路が考慮されます。パス1は、HサイトからSi原子の上部を横切る(Tサイトを横切る)隣接するHサイトへのLi空孔の移動を指します。パス2は、最上層のC2–C2ダイマーの中間点の上部を横切るパス(B2サイトを横切る)に対応します。パス3は、ダウンレイヤー(B1サイト全体)のC2–C2ダイマーに沿ったパスです。最適化された経路に沿ったエネルギープロファイルを図8bに示します。見られるように、これらの経路に沿ったLiイオン移動エネルギー障壁は、特にPath-3(0.052 eV)では非常に低くなっています。パス1とパス2に沿ったエネルギープロファイルは、1つのLi空孔が作成されたときにLiイオンが大きく緩和されるため、わずかに非対称です。パス3はB1サイト(エネルギー的に最も好ましい吸着サイト)を通過するため、パス3に沿った非常に低いエネルギー障壁は合理的です。ただし、Path-3だけでは、P-Si 2 の表面に完全なLi拡散ネットワークを形成することはできません。 C 4 。したがって、Path-1またはPath-2は拡散プロセスに参加する必要があり、希薄なLi空孔移動の全体的なエネルギー障壁は0.155eVまたは0.165eVです。希薄Liイオン移動(0.117 eV)よりも高いものの、希薄Li空孔移動のエネルギー障壁もP-C 6 の場合と比較して非常に小さいです。 (0.25 eV、Path-II ’)[21]およびg-Si 2 C 4 (0.233 eV)[13]。 P-Si 2 のLi移動エネルギー障壁として C 4 P-C 6 よりも常に低い およびg-Si 2 C 4 (希薄Liイオンと希薄Li空孔の両方)、P-Si 2 のレート性能が期待されます C 4 3つの類似したアノード候補の中で最高のものです。
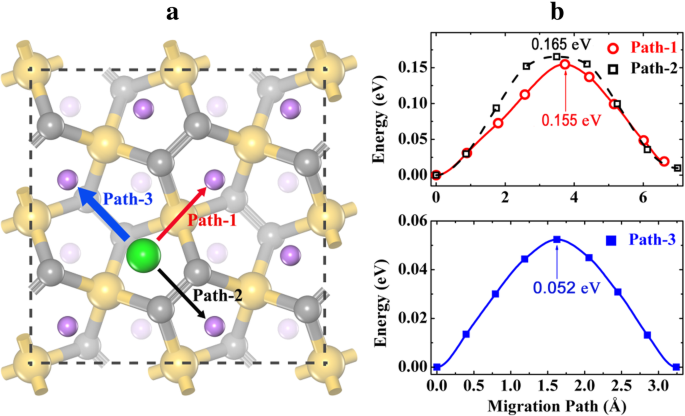
a Li空孔の移動経路を希釈して b 完全にリチウム化されたP-Li 4 の対応するエネルギープロファイル Si 2 C 4 。灰色(中型)、金色、紫色の球は、それぞれC、Si、Li原子です。大きな緑色の球は、希薄なLi空孔を表しています。太い/細い矢印は、2次元拡散ネットワークを形成する速い/遅い移動経路を示しています
結論
要約すると、第一原理計算に基づいて、2D五角形Si / C化合物P-Si 2 C 4 LIBのアノード材料として使用できる可能性があります。フォノン分散データにより、P-Si 2 の動的安定性が確認されました。 C 4 基底状態での構造、AIMDシミュレーションはP-Si 2 の構造を示しています C 4 2000 Kの高温でも安定します。独自の2D座屈五角形構造により、特別な空のC-2 p が促進されます。 z P-Si 2 の表面へのLiの吸着を促進する状態 C 4 、1028.7 mAhg -1 の重量分析Liストレージ容量を提供します 。計算された希薄Liイオン/ Li空孔移動エネルギー障壁は、P-Si 2 の表面でのLiイオン拡散を示しています。 C 4 五角形のグラフェン(P-C 6 )およびハニカム構造のシリグラフェン。リチウム化されたP-Li x の金属電子構造 Si 2 C 4 電極としての材料の良好な電子伝導性を保証します。これらの利点は、P-Si 2 にとって重要な機能です。 C 4 LIBの有望なアノード材料として。要約すると、私たちの第一原理研究は、LIB内のアプリケーションの高性能Si / Cの複雑さを設計するための新しい戦略を提供します。
データと資料の可用性
この調査中に生成または分析されたすべてのデータは、この公開された記事に含まれています。
略語
- 2D:
-
二次元
- AIMD:
-
Abinitio分子動力学
- bcc :
-
体を中心とした顔
- CINEB:
-
クライミング画像のナッジ輪ゴム
- DFT:
-
密度汎関数理論
- DOS:
-
状態密度
- EV:
-
電気自動車
- GGA:
-
一般的な勾配近似
- HSE06:
-
Heyd-Scuseria-Erznerhof
- LIB:
-
リチウムイオン電池
- PAW:
-
プロジェクター拡張波
- PBE:
-
Perdew-Burke-Ernzerhof
- SOC:
-
充電状態
- VASP:
-
ウィーンabinitioシミュレーションパッケージ
- vdW:
-
ファンデルワールス
ナノマテリアル
- Scalmalloy:金属3D印刷用の最新の高性能素材
- 将来のバッテリーのためのスズナノ結晶
- リチウムイオン電池用の高性能アノード材料としてMWNTに固定されたSiO2 @ Cナノ粒子の容易な合成
- リチウムイオン電池用の効率的なアノード材料としての数層のMoS2 /アセチレンブラック複合材料
- PPy被覆MnO2ハイブリッドマイクロ材料の調製とリチウムイオン電池のアノードとしてのそれらの改善されたサイクル性能
- リチウムイオン電池用の金属酸化物アノードの電気化学的性能に及ぼす異なるバインダーの影響
- 水性ナトリウムイオン電池用の高電気化学的性能材料としてのNa4Mn9O18 /カーボンナノチューブ複合材料
- リチウムイオン電池のアノード材料としてのマグネシウム-熱還元によって製造された埋め込みSi /グラフェン複合材料
- リチウムイオン電池用の高性能フレキシブルカソードとしてシード支援水熱プロセスを介して炭化繊維上に成長した3D相互接続V6O13ナノシート
- リチウムイオン電池用のパルスレーザー堆積によって調製されたナノ結晶Fe2O3膜アノード
- 高度なナトリウムイオン電池のアノード材料としてのCuGeO3ナノワイヤの合成と調査