


有機リンおよびカルシウム源は、高比表面積を有するリン酸カルシウムの卵黄殻構造ミクロスフェアの合成を誘導する:HEL吸着への応用
要約
卵黄殻構造のリン酸カルシウムミクロスフェアは、その優れた物理化学的特性と生体適合性により、医療用途に大きな可能性を秘めています。しかし、吸着能力の高い卵黄殻構造のリン酸カルシウムを開発することは依然として課題です。ここでは、比表面積の高いリン酸カルシウムの多孔質卵黄殻構造ミクロスフェア(ATP-CG)[ S ベット =143 m 2 g -1 カルシウム源をグルコン酸カルシウム五水和物(CL)に置き換えて合成したATP-CLミクロスフェアの約3倍の高さ]は、アデノシン5'-三リン酸二ナトリウム塩(ATP)をリン源として使用することで合成に成功しました。自己テンプレートアプローチによるカルシウム源としてのグルコン酸カルシウム一水和物(CG)。 ATP-CGミクロスフェアの形態に及ぼすCa対Pのモル比(Ca / P)、熱水温度、および時間の影響も調べた。有機カルシウム源と有機リン源が卵黄殻構造の形成に重要な役割を果たしていることがわかった。さらに、異なるカルシウム源で合成された2種類の卵黄殻構造のミクロスフェアの吸着メカニズムを明らかにするために、一連の吸着実験が調査されました。結果は、ATP-CGミクロスフェア(332±36 mg / g)の吸着容量がATP-CLミクロスフェア(176±33 mg / g)の吸着容量の約2倍であることを示しています。さらに、カルシウム源によって引き起こされるより高い比表面積およびATP-CGミクロスフェアの独特の表面化学的性質は、HEL吸着能力の改善において重要な役割を果たします。この研究は、調製されたままの卵黄殻構造のミクロスフェアがドラッグデリバリー分野での応用に有望であり、薬物吸着能力を改善するための効果的なアプローチを提供することを示しています。
はじめに
リン酸カルシウムは、その優れた生体適合性[1]、高い負荷容量、および送達効率により、過去数年間でかなりの注目を集めています。リン酸カルシウム関連の生体材料は、組織工学[2]、骨修復[3]、ドラッグデリバリー[4]などのさまざまな生物医学分野で広く使用されています。リン酸カルシウムベースの材料の適用範囲を拡大し、性能を向上させるために、炭酸ヒドロキシアパタイト(HAp)ミクロスフェア[5]、HApマイクロチューブ[6]、中空HApミクロスフェア[7]などのさまざまな形態と微細構造を持つさまざまなリン酸カルシウム材料]、アモルファスリン酸カルシウム(ACP)のメソポーラスyolk @ cage-shellナノスフェア[8]が報告されています。
さまざまな形態の中で、卵黄殻構造のミクロスフェアは、フロンティア材料科学であるだけでなく、独特の形態的特徴を示すため、ますます注目を集めています。卵黄殻構造のミクロスフェアでは、卵黄のコアと殻の間の空間がさまざまな貨物の貯蔵庫として機能し、多孔質構造の殻がゲスト分子の拡散経路を提供できるため、次のようなさまざまな用途に大きな可能性を秘めています。触媒作用[9]、リチウムイオン電池[10]、光触媒[11]、および生物医学[12]。伝統的に、卵黄殻構造のミクロスフェアを調製するための主要な方法は、テンプレート法を犠牲にすることです[13、14]。これらのテンプレート戦略は、構造とプロパティの調整に大きな成功を収めています。ただし、これらのアプローチにはいくつかの欠点があります。たとえば、面倒な処理ステップや、人間の健康に害を及ぼす可能性のある界面活性剤や構造指向試薬などです。現在、自己テンプレート法は、卵黄殻構造のミクロスフェアの研究に広く使用されています[15、16]。従来のテンプレートアプローチとは異なり、自己テンプレートアプローチで使用されるテンプレートは、ボイドを形成するためのテンプレートであるだけでなく、卵黄殻構造のミクロスフェアの前駆体でもあります。したがって、自己テンプレート法は、卵黄殻構造のミクロスフェアを調製するための便利なアプローチです。ただし、卵黄殻構造のリン酸カルシウムミクロスフェアの合成に自己テンプレートアプローチを導入することは、依然として興味深い課題です。
さらに、リン酸カルシウム材料は、タンパク質[17]、DNA [18]、siRNA [19]などのさまざまな種類の貨物を運ぶために利用されてきました。ただし、リン酸カルシウムの薬物吸着能力の低さは早急に解決する必要があります。一般に、担体の表面に固定化する薬物分子のアプローチは、表面電位[20]、疎水性/親水性[21]、水素結合[22]、および比表面積[23]を含む表面特性に依存します。したがって、表面特性と比表面積を改善することは、担体の薬物吸着能力を高めるための有効なアプローチです。
ここでは、自己テンプレートアプローチにより、リン酸源としてアデノシン5'-三リン酸二ナトリウム塩(ATP)を、カルシウム源としてグルコン酸カルシウム一水和物(CG)を使用して、リン酸カルシウムの一種の多孔質卵黄殻構造ミクロスフェアを調製しました。テンプレート剤を添加しなくても、調製されたままの卵黄殻構造のリン酸カルシウムミクロスフェアは、特に高い比表面積を示します。さらに、ATP-CGミクロスフェアの鶏卵リゾチーム(HEL)吸着挙動を、カルシウム源をカルシウムl-乳酸五水和物(CL)に置き換えて調製したATP-CLミクロスフェアと比較して調査しました。この結果は、カルシウム源と表面の化学的性質によって引き起こされる比表面積の違いが、HEL吸着能力の向上に重要な役割を果たしていることを示しています。
メソッド
資料
アデノシン5'-三リン酸二ナトリウム塩(ATP)は、Macklin Biochemical Co.、Ltd(Shanghai、China)から入手しました。グルコン酸カルシウム一水和物(CG)および乳酸カルシウム(l)-乳酸五水和物(CL)は、Sangon Biotech Co.、Ltd(Shanghai、China)から購入しました。鶏卵リゾチーム(HEL、〜70000 U / mg)は、Sigma-Aldrich(タウフキルヘン、ドイツ)から購入しました。
ATP-CGおよびATP-CL卵黄殻構造ミクロスフェアの合成と特性評価
ATP-CGヨークシェル構造のリン酸カルシウムミクロスフェアは次のように調製しました。簡単に説明すると、0.9gのCGを20mLの超純水に溶解して60°Cで溶液Cを形成し、0.11gのATPを5mLに溶解しました。次に、溶液Cを室温まで冷却し、激しく攪拌しながら溶液Pと混合し、溶液のpHを2 MNaOH溶液で5に調整しました。溶液の最終容量は30でした。超純水を追加したmLで、CaとPのモル比(Ca / P)は3.3でした。最終溶液をマイクロ波水熱反応用のマイクロ波消化システムに移し、120°Cで15分間処理しました。得られた沈殿物を遠心分離(4500rpm、10分)によって収集し、超純水ですすぎ、48時間凍結乾燥した。 ATP-CLミクロスフェアは文献の手順[24]に従って調製されました。
ミクロスフェアの結晶相は、X線回折(XRD、CuK α)によって特徴づけられました。 ソース、λ =0.154)。ミクロスフェアの形態は、走査型電子顕微鏡(SEM)、透過型電子顕微鏡(TEM)、および高分解能TEM(HRTEM)によって観察されました。ミクロスフェアの組成は、フーリエ変換赤外分光光度計(FTIR)によって研究されました。ミクロスフェアの比表面積は、Brunauer-Emmett-Teller(BET)によって決定されました。熱重量分析(TGA)を使用して、窒素雰囲気中で10°C /分の加熱速度でサンプルの熱特性を研究しました。
HELの吸着と特性評価
2種類のミクロスフェアのHEL吸着実験は、次のように実施されました。一定量の卵黄殻ミクロスフェア(ATP-CG、Ca / P =3.3、120°C、15分、およびATP-CL、Ca / P =2.5、 120°C、30分)を10分間の一定の超音波処理で水に分散させ、1.5 mg / mLのミクロスフェア懸濁液を形成しました。次に、さまざまな濃度のHELを含む0.5 mLの水溶液を、懸濁液の上の1 mLにすぐに添加し、薬物の最終濃度は1〜7.5 mg / mLでした。各溶液を37°Cで6時間振とうしました(200rpm)。その後、溶液を遠心分離し、上澄み中のHELの量を280nmでUV-vis分光光度計によって測定しました。薬物負荷前後のゼータ電位とミクロスフェアの組成は、ゼータ電位分析装置、FTIR分光計、および熱重量分析装置(TGA、加熱速度10°Cmin -1 )によって特徴付けられました。 、窒素雰囲気)。
吸着等温線
吸着挙動を調査するために、Dubinin-Radushkevic等温線(D-R)モデルを本研究で実施しました。 D-Rモデルは、ミクロポア充填の理論に基づいています。これは、不均一な表面での非理想的な収着を説明し、収着メカニズム(物理的収着または化学的収着)を区別するために使用されます。モデルは次の方程式で表されます。ここで、 Q eq は平衡状態での吸着剤の吸着容量(mg / g)、 C eq は平衡状態の水相の吸着質濃度(mL / L)です。 Q m 最大吸着容量です。 R は気体定数、8.314 J /(mol∙k)です。 T は絶対温度です。 E 吸着のタイプを推定するための平均自由エネルギーを表します。 E の場合 値が8kJ / mol未満の場合、吸着タイプは物理吸着で説明できます。8〜16 kJ / mol、吸着タイプはイオン交換に属し、16 kJ / molを超える場合、吸着タイプは化学吸着で説明できます。 。
$$ {Q} _ {\ mathrm {eq}} ={Q} _m \ exp \ left(-{K} _ {\ mathrm {DR}} \ {\ varepsilon} ^ 2 \ right)$$(1) $$ \ varepsilon =\ text {RT1n}(1+ \ frac {1} {{C} _ \ text {eq}})$$(2)$$ \ mathrm {E} =\ frac {1} {\ sqrt {K _ {\ mathrm {DR}}}} $$(3)薬物吸着の統計分析
データは平均±標準偏差(SD)値として表されました。重要な違い( p <0.05)は、一元配置分散分析を使用して、異なるグループ間で統計的に計算されました。すべての実験は3回実施され、データはDPSソフトウェアを使用して分析されました。
結果と考察
ミクロスフェアの形態と化学的特性評価
ATP-CG卵黄-殻構造のミクロスフェア
図1のSEM画像は、さまざまな反応条件下で得られたさまざまなサンプルの形態を示しています。 t で =5分または15分、すべての製品は均一なミクロスフェアで構成されています。しかし、熱水時間をさらに30分に増やすと、ナノシートの自己組織化ミクロスフェアが形成されました(図1 f、i、lに示すように)。一方、製品の形態に対するCa / Pの影響は、 t でも観察されます。 =30分Ca / Pの増加に伴い、ナノシートの自己組織化ミクロスフェアが徐々に形成されました(図1f、i、lに示すように)。ナノシートの自己組織化ミクロスフェアの形成は、以下の理由で説明できます。まず、マイクロ波熱水プロセスでは、ATP分子が加水分解して、アデノシン二リン酸(ADP)、アデノシン一リン酸(AMP)、アデノシンなどのアデノシンベースの分子を形成し、同時にリン酸イオンを放出します(PO 4 3- )。一方、CG分子は加水分解して、グルコン酸塩とカルシウムイオン(Ca 2+ )を形成する可能性があります。 )。次に、リン酸イオンはカルシウムイオンと反応して一次ACP核を形成します[25]。次に、最初のACP核が成長して集合し、ACPミクロスフェアを形成します。したがって、熱水時間がさらに延長されると、溶液中のATPおよびCG分子はさらに加水分解され、より多くのPO 4 を放出します。 3- およびCa 2+ イオンは、システムの過飽和と核形成速度を改善することにより、ナノシートの自己組織化ミクロスフェアの形成を引き起こします。さらに、Ca / Pを増やすことにより、Ca 2+ の局所的な高濃度 また、上記と同じ方法で製品の形態変換を加速します。上記の分析は、熱水時間とCa / Pが製品の形態に重要な影響を与えることを示しています。

120°Cでマイクロ波水熱法によって調製されたATP-CGミクロスフェアのSEM画像
次に、さまざまなCa / Pを使用して120°Cで15分間合成したミクロスフェアのFTIRスペクトルを調べます(図2)。 1620 cm -1 のピーク 、1383 cm -1 、および912 cm -1 ATPのCGおよびP–OグループのC =O、C–Oの特徴的なピークにそれぞれ起因し[26]、加水分解されていないCGおよびATP分子またはそれらの誘導体がミクロスフェアの表面に吸収されることを意味します。 PO 4 のかすかな特徴的なピーク 3- HApからは1035cm -1 にあります [27]および1122cm -1 での吸収ピーク と567cm -1 PO 4 に割り当てられます 3- ACPのイオン[28]は、製品がACPとHApで構成されていることを示しています。 FTIRの結果は、リン酸カルシウムがリン源としてATPを使用し、カルシウム源としてCGを使用することによって正常に調製されたことを示唆しています。
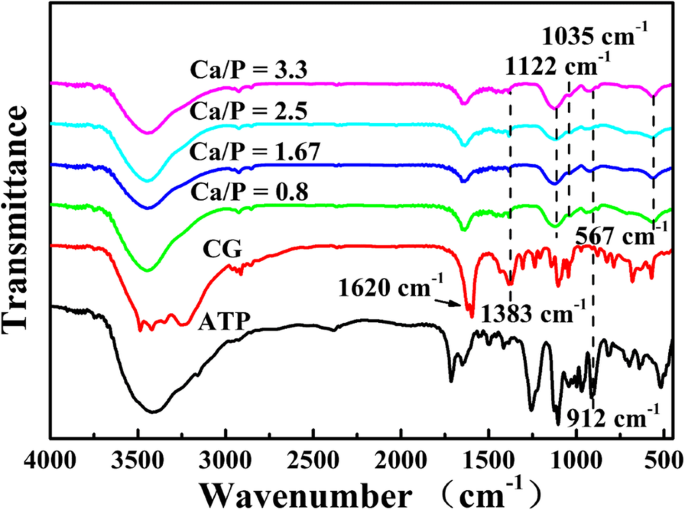
120°Cで15分間さまざまなCa / Pで合成されたATP-CGミクロスフェアのFTIRスペクトル
さらに、120°Cで15分間のマイクロ波水熱法によりさまざまなCa / Pで合成されたサンプルのSEMおよびTEM画像を図3に示します。Ca/ Pが0.8または1.67の場合、サンプルは多孔質ミクロスフェアで構成されます(図3b、d)。 Ca / Pが2.5になると、生成物の形態が卵黄殻構造のミクロスフェアに変化し始めます(図3f)。 Ca / Pがさらに3.3に増加すると、製品は完全に卵黄殻構造のミクロスフェアで構成されます(図3h)。それを超えて、いくつかの壊れた球と卵黄殻ミクロスフェアの露出したコア(図3gに挿入)は、機械的破砕後に観察され、卵黄と殻の間に中空構造の証拠を提供します。以上の観察を踏まえ、様々なCa / Pで合成された卵黄殻構造のミクロスフェアの形成メカニズムを暫定的に提案する。 Ca / Pが低い場合、最初に多孔質ACPミクロスフェアが形成されます。これは、ミクロスフェアの表面に吸着されたATPおよびCG分子またはそれらの誘導体の抑制効果に起因します。次に、Ca / Pがさらに増加すると、準安定ACPがさらに成長します。これは、システムの高い過飽和によって駆動されます。最後に、結晶性HApが外面に形成されます。これは、図3iのミクロスフェアの高分解能TEM(HRTEM)画像によって確認されます(0.308 nmの面間距離はHApの(210)にインデックス付けできます)。その結果、HApとACPの体積または密度の違いにより、卵黄と殻の間に中空構造が生成されます[24]。対応するEDSマッピングは、Ca、P、およびO元素がミクロスフェア全体に均一に分布していることを示しています。図3kのEDSスペクトルと図3lのXPSスペクトルは、ミクロスフェアの化学元素が主にCa、P、およびOを含むことを示しており、FTIRの結果と一致しています(図2)。

さまざまなCa / Pで合成されたATP-CGミクロスフェアのSEMおよびTEM画像。 a 、 b Ca / P =0.8。 c 、 d Ca / P =1.67。 e 、 f Ca / P =2.5。 g 、 h Ca / P =3.3。 i HRTEM、 j EDSマッピング、 k EDSスペクトル、 l Ca / P =3.3
のATP-CGミクロスフェアのXPSスペクトルCa / P =3.3で合成されたミクロスフェアの形態に対するマイクロ波支援熱水時間と温度の影響をさらに調査します。図4a-bに示すように、熱水時間が5分である場合、サンプルは多孔質ミクロスフェアで構成されます。上で説明したように、 t =15分、製品も卵黄殻構造のミクロスフェアで構成されています(図4c-d)。熱水時間が60分に延長された場合、または温度が160 o に上昇した場合 C、シートまたはロッドの束縛が観察されます(図4e-l)。多孔質から卵黄殻、シートまたはロッドへの形態変化は、溶液中のATPおよびCG分子の連続加水分解によるACPのさらなる成長に起因します。さらに、ACPミクロスフェアの表面に吸着されたATPおよびCG分子またはそれらの誘導体の加水分解もACPの成長を加速します。 60分または160 o で興味深い現象が発生しました C、これらのシートまたはロッドもACPナノ粒子(赤いボックスで示されている)から開発されています。これは、図S1のDTA分析によって確認されています。 650°Cでの発熱ピークがDTA曲線で観察され[29、30]、これはACP結晶化に起因します。発熱ピークは、熱水時間または温度の上昇とともに徐々に弱くなり、生成物中のACPがリン酸カルシウムクリスチリンに変換されることを意味します。

さまざまな実験条件下でCa / P =3.3で合成されたATP-CGミクロスフェアのSEMおよびTEM画像。 a 、 b T =120°C、 t =5分。 c 、 d T =120°C、 t =15分。 e–h T =120°C、 t =60分 i–l T =160°C、 t =15分
異なる熱水時間または温度の下でCa / P =3.3で合成されたサンプルの化学構成と構造は、FTIRとXRDによって調査されます。図5aに示すように、PO 4 の特徴的なピーク 3- HApのイオンは1037cm -1 にあります および603cm -1 [27]。 1122 cm -1 のピーク PO 4 の特徴的なピークに割り当てられます 3- ACPからのイオン。吸収のピークは1620cm -1 および1383cm -1 CGからのC =OおよびC–Oグループの特徴的なピークにそれぞれ起因します。 912 cm -1 の吸収ピーク ATPの非対称P–O伸縮振動を指します。熱水時間または温度を上げることにより、CGおよびATPの特徴的なピークの強度が徐々に減少し、ミクロスフェアの表面に吸着されたATPおよびCG分子またはそれらの誘導体がさらに加水分解されることを示します。一方、PO 4 の特徴的なピークの強度 3- HApのイオンは、ACP特性ピークの強度が低下するにつれて徐々に増加する傾向を示し、生成物の結晶相からHAp相への変換を明らかにします。
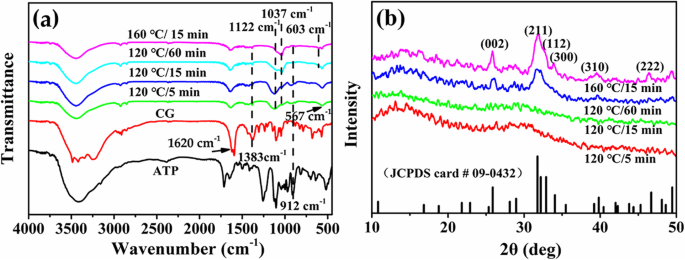
a FTIRスペクトルと b さまざまな実験条件下でCa / P =3.3で合成されたATP-CGミクロスフェアのXRDパターン
図5bは、さまざまなサンプルのXRDパターンを示しています。 2 θ付近のアモルファス相の特徴的なこぶ =5分または15分で合成された30°のミクロスフェアが観察されます。ただし、熱水時間を60分に延長するか、温度を160°Cに上げると、ミクロスフェアの結晶相は完全にHApに変化し、標準データとしてインデックス化できます(JDCPS番号09-0432)。 (211)、(300)、および(002)格子面の相対強度の改善は、製品の結晶化度の増加をさらに説明する可能性があります。したがって、XRDおよびFTIRの結果は、熱水温度または時間の増加に伴う生成物の結晶相変態をさらに確認します。
ATP-CL卵黄-殻構造のミクロスフェア
薬物吸着挙動を比較するために、他の卵黄殻構造のミクロスフェアは、マイクロ波水熱法によって有機カルシウム源としてCLを使用して調製されました[24]。形態に関しては、サンプルはまだ卵黄殻構造のミクロスフェアで構成されており、これは壊れたスフィア(図6aに挿入)とTEM画像(図6b、c)によって確認されます。この結果は、カルシウム源の変化が製品の形態に有意な影響を及ぼさないことを示しています。さらに、選択領域電子線回折(SAED)は、個別のSAEDスポットを示し(図6d)、十分に結晶化したミクロスフェアが得られることを示しています。さらに、EDSマッピングは、ミクロスフェア内のCa、P、およびO元素の均一な分布を示します(図6e)。対応するEDSスペクトルは、ミクロスフェア内のCa、P、およびOエレムネットの存在も確認し(図6f)、準備されたままのミクロスフェアがリン酸カルシウムであることを示しています。

a SEM 。 b 、 c TEM画像。 d S 選択領域電子回折(SAED)。 e EDSマッピングと f ATP-CLミクロスフェアのEDSスペクトル
HEL吸着およびミクロスフェアの吸着メカニズム
図7に示すように、2種類のミクロスフェアの吸着容量は、HELの初期濃度の増加とともに増加します。 HELの初期濃度が6.5mg / mLに増加すると、ATP-CGミクロスフェアの吸着容量はプラトーに達し、ミクロスフェアの最大吸着容量は約332±36 mg / g(図7a)であり、これは約2倍です。 ATP-CLミクロスフェアよりも高い(176±33 mg / g、6 mg / mL、図7b)。
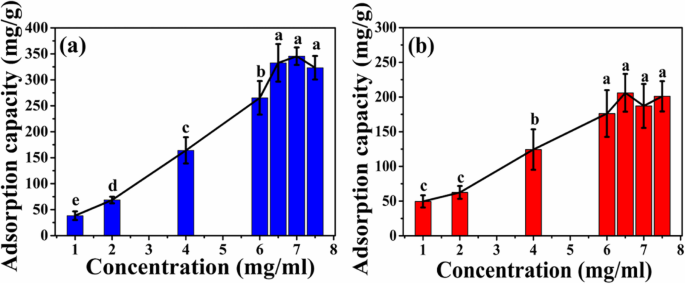
HELの異なる初期濃度でのミクロスフェアの吸着曲線。 a ATP-CGミクロスフェア。 b ATP-CLミクロスフェア
HEL吸着の結果は、FTIRスペクトルとTG曲線によってさらに裏付けられます。図8a、bに示すように、1134 cm -1 に吸収ピークがあります。 (1139 cm -1 )および563 cm -1 (568 cm -1 )特徴的なピークPO 4 に割り当てられます 3- ACPおよび1039cm -1 のイオン (1040 cm -1 )PO 4 の特徴的なピークに割り当てられます 3- HApのイオンは、HELに吸着されたミクロスフェアで観察されます。これは、ミクロスフェアにHELを導入しても、ミクロスフェアの構造に大きな変化が生じないことを示しています。吸着のピークは1542cm -1 および1545cm -1 HELのアミド基に起因するものがHEL吸着ミクロスフェアで観察され、HELがミクロスフェアにうまく吸着されていることが確認されます。一方、2966、2962、2935、および2927 cm -1 の吸着バンド –CH 3 に由来 および–CH 2 HELのグループは、HELに吸着されたミクロスフェアでも検出され、ミクロスフェア上のHELの存在をさらに検証します。 TGA曲線は、HEL吸着前後のATP-CGミクロスフェアの重量損失がそれぞれ11.3%と36.7%であることを示しています(図8c)。したがって、ATP-CGミクロスフェアのHEL吸着容量は約340mg / gです。しかし、HEL吸着前にATP-CLの21.1%の重量損失が得られ、37%がHEL吸着ミクロスフェアに現れます(図8d)。したがって、ATP-CLミクロスフェアのHEL吸着容量は189 mg / gです。 TGAの結果は、図7の結果に近いものです。
HEL吸着前後のミクロスフェアのFT-IRスペクトルとTGA曲線。 a FTIRスペクトルと c ATP-CGミクロスフェアのTGA曲線、 b FTIRスペクトルと d ATP-CLミクロスフェアのTGA曲線
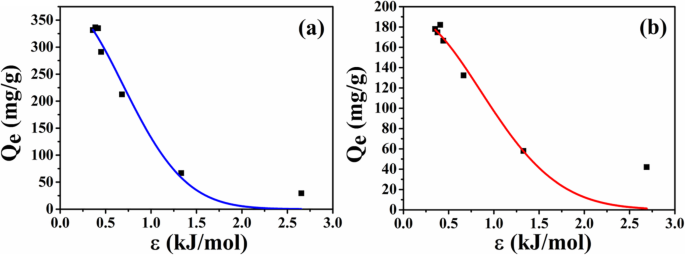
2種類のミクロスフェア間の吸着容量の違いの原因を調査するために、ミクロスフェアの平衡吸着データをD-R等温線モデルに従ってさらに分析します。フィッティング曲線を図9に、フィッティングパラメータを表1にそれぞれ示します。フィッティング結果から、ATP-CGからの相関係数はATP-CLよりも高く、D-RモデルがATP-CGミクロスフェアの薬物吸着挙動を記述するのに適していることを示唆しています。 E 以降 値が8kJ / mol未満の場合、ATP-CGミクロスフェアへのHELの吸着は物理的吸着です。最大容量( Q m )HELのATP-CGミクロスフェアは381 mg / g近くに達する可能性があり、これは図7aの結果に近いものです。
a ATP-CGミクロスフェア上のHELの吸着等温線モデル。 b ATP-CLミクロスフェア上のHELの吸着等温線モデル
ATP-CGミクロスフェアへのHELの吸着は物理的吸着であるため、ミクロスフェアの表面電位を調査します。図10aに示すように、超純水中のATP-CG、ATP-CLミクロスフェア、およびHELのゼータ電位値は、それぞれ– 17 mV、– 22 mV、および20mVです。 HEL吸着後、ATP-CGおよびATP-CLミクロスフェアのゼータ電位値はそれぞれ2.7mVおよび1.5mVに変化し、引力による静電力によるミクロスフェアの表面へのHEL分子の吸着を示しています。ただし、ミクロスフェア間でゼータ電位値(-17 mVと– 22 mV)に大きな違いがないため、引力の静電力は2種類のミクロスフェア間の吸着容量の違いの主な原因ではありません。
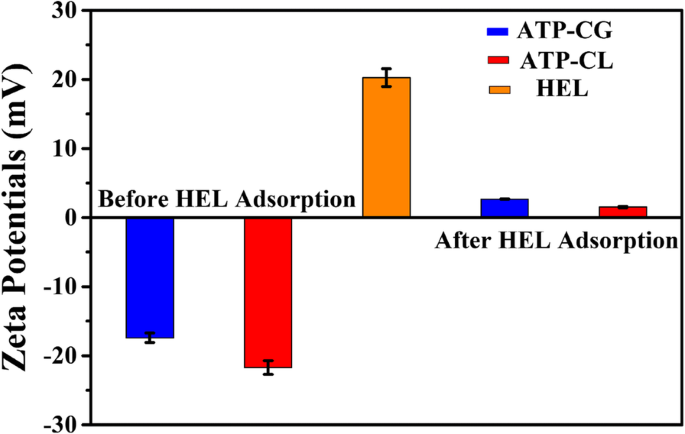
HEL吸着前後のHELとミクロスフェアのゼータ電位
したがって、ミクロスフェア間の吸収能力の差を引き起こす理由をさらに明らかにするために、ミクロスフェアの比表面積を調査する。図11aに示すように、BET比表面積( S ベット )ATP-CGミクロスフェアの量は143 m 2 g -1 、ATP-CLミクロスフェアの約3倍の高さ(55 m 2 g -1 、表2)。したがって、特定の表面積は、ミクロスフェア間の吸収能力の違いに寄与する可能性があります。 ATP-CGミクロスフェアのこのような高比表面積は、主に結晶化度が低いことに起因します[31]。図11bから、ATP-CGミクロスフェアはATP-CLミクロスフェアよりも低い結晶化度を示します。さらに、ATP-CGとATP-CLの結晶化度の違いは、主に合成条件の違いによるものです。一般に、生成物の結晶化度は、特定の圧力と温度での反応物の加水分解の程度とともに増加します。ここで、グルコン酸(pKa =3.39)の酸性度はl-乳酸(pKa =3.86)よりも高く、これにより加水分解速度が遅くなり、最終的に結晶化度が低くなります。その結果、カルシウム源を追いかけることで、比表面積の大きいATP-CGミクロスフェアが得られます。
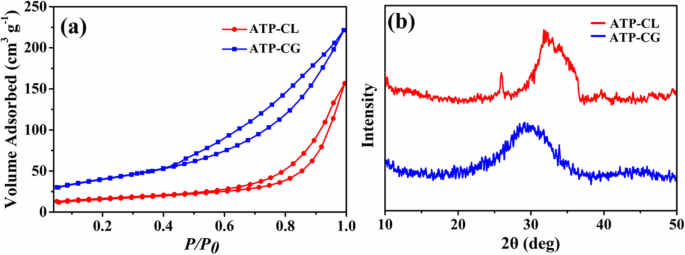
a 窒素の吸脱着等温線。 b ミクロスフェアのXRDパターン
結論
ATP-CG卵黄殻ミクロスフェアは、有機リン源としてATPを使用し、有機カルシウム源としてCGを使用することにより、マイクロ波支援水熱法によって設計されています。ミクロスフェアは、高い比表面積と高い吸着能力を示します。ミクロスフェアの形態と構造に対するCa / P、熱水温度、および時間の影響も調査されました。この研究は、有機リン源と有機カルシウム源が卵黄殻構造のミクロスフェアの形成に大きな影響を与えることを示しています。さらに、Ca / P、熱水、および温度を含む熱水条件は、卵黄殻ミクロスフェアの形成に関与しています。さらに、異なるカルシウム源で合成された2種類のミクロスフェアのHEL吸着挙動を比較することにより、比表面積と表面電位などの表面化学的特性がミクロスフェアの吸着容量に影響を与える2つの重要な要因であることがわかります。
データと資料の可用性
この記事の結論を裏付けるすべてのデータが記事に含まれています。
略語
- ベット:
-
Brunauer-Emmet-Tellerの測定値
- FTIR:
-
フーリエ変換赤外分光法
- TEM:
-
透過型電子顕微鏡
- XRD:
-
X線回折
- TGA:
-
熱重量分析
- HRTEM:
-
高分解能TEM
- SAED:
-
選択領域電子回折
ナノマテリアル
- ICAをロードしたmPEG-ICAナノ粒子の調製とLPS誘発性H9c2細胞損傷の治療におけるそれらの応用
- 超狭帯域完全吸収体と可視領域のプラズモニックセンサーとしてのその応用
- CuSナノ粒子でコーティングされた着色および導電性CuSCN複合材料の容易な合成
- MnХFe3−XО4スピネルの構造的および磁気的特性に及ぼす接触非平衡プラズマの影響
- TIPS-ペンタセンベースの有機電界効果トランジスタの移動度と形態に及ぼすその場アニーリング処理の影響
- 界面層の設計によるZnO膜の表面形態と特性の調整
- 表面および層間の修飾による油中の層状リン酸ジルコニウムナノプレートレットのトライボロジー性能の調整
- ZnOナノ結晶の合成と逆ポリマー太陽電池への応用
- ポリ[(9,9-ジオクチル-2,7-ジビニレンフルオレニレン)-alt-co-(2-メトキシ-5-(2-エチルヘキシルオキシ)-1,4-フェニレン)](POFP)のレーザー発振および輸送特性ダイオード励起有機固体レーザーの応用
- メタマテリアルにおける表面プラズモンポラリトンと磁気双極子共鳴の結合効果
- アップコンバージョン発光を増強するための異なる形態のBaYF5:Er3 +、Yb3 +の制御された合成