


骨髄間葉系幹細胞由来のエクソソームマイクロRNA-133aは、MAML1を抑制することにより、ウイルス性心筋炎ラットの心筋線維化と上皮間葉転換を抑制します。
要約
心筋炎は、効率的な治療を行わずに心筋の限局性またはびまん性の炎症を特徴とする疾患です。この研究では、骨髄間葉系幹細胞由来のエクソソーム(BMSC-Exo)から分泌されるmicroRNA-133(miR-133)が、心筋炎およびウイルス性心筋炎(VMC)ラットの上皮間葉転換(EMT)で調節することにより、調節メカニズムを調べました。首謀者のような1(MAML1)。ラットのBMSCを分離および培養して、免疫表現型と骨形成および脂肪生成能力を特定し、BMSC-Exoを抽出して特定しました。エクソソームは超遠心分離によって得られ、透過型電子顕微鏡法とウエスタンブロット分析によって同定されました。ラットにコクサッキーB3ウイルスを注射して、VMCモデルを作成し、動物実験と同じ方法で心筋細胞を分離、培養、グループ化しました(NC Exo 、Ad-miR-133a Exo 、Adas-miR-133a Exo )。炎症、アポトーシス、EMT、線維症、および細胞生存率におけるエクソソームmiR-133aおよびMAML1の役割を解明するために、invivoおよびinvitro実験が実施されました。 miR-133aとMAML1の間のターゲティング関係は、デュアルルシフェラーゼレポーター遺伝子アッセイによって検証されました。 BMSC-ExoはVMCラットでmiR-133aの発現を上昇させ、VMCラットの心機能と心筋線維症を効果的に改善し、心筋細胞の生存率を高め、EMTプロセスを阻害しました。エクソソームのmiR-133aの上昇は、改善を強化しました。沈黙したmiR-133aは、VMCラットに対するBMSC-Exoの効果を効果的に逆転させました。 miR-133aはMAML1をターゲットにしました。 MAML1の阻害は、VMCラットの心機能と心筋線維症を改善し、VMCラットに対するmiR-133aサイレンシングされたエクソソームの効果を逆転させる可能性があります。私たちの研究は、エクソソームmiR-133aの上昇が、MAML1のダウンレギュレーションを介して、VMCのラットの心筋線維化とEMTを抑制し、それによって心筋炎の進行を抑制することを示唆しています。
はじめに
心筋炎は、心筋細胞の炎症性疾患と見なされています[1]。心筋炎は、女性とは対照的に男性の方が明らかによく知られています[2]。ウイルス性心筋炎(VMC)は、拡張型心筋症(DCM)と若者の突然死につながる主な要因です[3]。心筋炎の臨床成績は、兆候や症状があいまいな無症候性の状態から、心原性ショックや不整脈に苦しむウイルスや免疫細胞による深刻な心筋破壊まで、さまざまです[1]。心筋炎は、ウイルス、細菌、クラミジア、リケッチア、真菌、原生動物などのさまざまな感染性要素と、非感染性の誘導物質によって誘発される可能性があります。その中で、特に子供では、ウイルス感染が最も一般的な原因でした[4]。コクサッキーB3ウイルス(CVB3)は、心筋炎を引き起こす最も重要なウイルスとして、VMCの病因に酸化ストレス応答とアポトーシスを引き起こす可能性がありますが、VMCの特別な治療法はまだ報告されていません[5]。さらに、VMCの病因は十分に文書化されておらず、正確な臨床治療も不足しています[3]。したがって、疾患の予後を改善するために、新しい標的が緊急に必要とされています。
マイクロRNA(miRNA)は、タンパク質をコードする遺伝子の発現を調節できる内因性の非コードRNAです[6]。 MiR-133aは、心臓特異的miRNAの1つとして、心臓発作および心筋梗塞(MI)を含むいくつかの心血管疾患に関与しています[7]。さらに、miR-133aは、慢性シャーガス病の心筋症で異常に発現するものです[8]。さらに、心筋のmiR-133aレベルは、炎症、左心室機能、および炎症性心筋症の臨床転帰と関連しています[9]。 miRNAは、マウスおよびヒトの肥満細胞に由来するエクソソームで発見されました[10]。エクソソーム、ほとんどの細胞種によって放出されるナノサイズの小胞は、さまざまな体液に見られます[11]。エクソソームはその積荷をレシピエント細胞に移すことができ、これはレシピエント細胞の生化学的組成とシグナル伝達経路を変化させることが示されています[12、13]。証拠は、変化したエクソソームmiRNAがCVB3誘発性心筋炎の病因と関連していることを示しています[14]。低酸素状態の骨髄間葉系幹細胞からのエクソソームmiR-125b-5pは、心筋細胞のアポトーシスを減少させ、虚血性心臓修復を促進することが指摘されています[15]。さらに、MSCからのエクソソームmiR-25-3pは、心筋細胞のアポトーシスと炎症反応を軽減することでMIを軽減します[16]。興味深いことに、エクソソームで心臓に発現するmiR-133aは、心臓のトロポニン心臓トロポニン-Iに関連しています[17]。 Mastermind-like 1(MAML1)は、心筋虚血/再灌流(I / R)傷害に関与することが報告されている我々の研究におけるmiR-133aのクロススクリーニングされた下流遺伝子でした[18]。また、最近の研究では、MAML1ノックダウンが肝線維症において抗線維化機能を持っていることが言及されています[19]。
以前の研究に照らされて、BMSCs由来のエクソソームmiR-133aが心筋炎を媒介できるかどうか疑問に思います。したがって、この研究は、BMSC由来のエクソソーム(BMSC-Exo)によってシャトルされたmiR-133が、MAML1の調節を通じてVMCラットの心筋線維化と上皮間葉転換(EMT)を改善するという仮説から始まりました。
材料と方法
倫理的承認
この研究は、浙江大学医学部の第4付属病院の施設内動物管理使用委員会によって許可されました。動物は人道的に扱われました。
BMSCの分離
実験動物は、成熟した特定病原体除去(SPF)グレードのSprague-Dawley(SD)雄ラット(浙江大学医学部実験動物センター、浙江、中国)でした。ペントバルビタールナトリウムの腹腔内注射によりラットを安楽死させ、75%アルコールで滅菌した。大腿骨と脛骨を超清浄テーブルで取り出し、筋肉と結合組織を取り除き、骨髄腔を低グルコースのダルベッコ改変イーグル培地(DMEM)で繰り返しすすいだ。液体を遠心分離して沈殿物を収集し、これを再懸濁して24時間インキュベートしました(培地は2〜3日ごとに交換しました)。対数増殖期に成長したとき、BMSCを0.25%トリプシン(Gibco、カールスバッド、カリフォルニア、米国)で分離し、遠心分離し、MSC培養液(Cyagen Biosciences Inc.、広州、中国)に再懸濁しました。懸濁液を1:2の比率で継代した。上記の操作を繰り返し、4継代のBMSCをその後の実験に使用しました。
BMSCの識別
対数増殖における4継代BMSCの表面抗原は、フローサイトメトリーによって同定されました。 BMSCをエチレンジアミン四酢酸を含む0.25%トリプシン(1 mL)で分離し、遠心分離し、適切なリン酸緩衝生理食塩水(PBS)で再懸濁し、151gで遠心分離しました。次に、BMSCを2%の新鮮なウシ胎児血清(FBS)(Gibco)を含むPBSで再懸濁して、単細胞懸濁液を作成しました。 FITC-CD34、PE-CD29、およびPE-CD44モノクローナル抗体(各5μL、BD Biosciences、フランクリンレイクス、ニュージャージー州、米国)を細胞懸濁液(100μL)とインキュベートし、151 gで遠心分離し、500で再懸濁しました。 1%パラホルムアルデヒドを含み、30分間固定したμLPBS。バックグラウンドマーカーは、ホモタイプコントロールのモノクローナル抗体を使用して特定されました。
フローサイトメトリー:単細胞懸濁液を固定し、151gで遠心分離しました。次に、BMSCを1%パラホルムアルデヒドを含むPBSで再懸濁し、MACSクォートフローサイトメーターでテストし、対応するソフトウェアで分析しました。
BMSCの骨化および脂肪生成の誘導
4継代のBMSCを200細胞/ mLの6つのウェルプレートに播種しました。骨芽細胞誘導溶液と脂肪生成誘導溶液(Cyagen Biosciences Inc.)を、60〜70%のコンフルエンスのBMSCに添加しました。他の2つのウェルのBMSCには、コントロールとして誘導液を添加しませんでした。 BMSCは14日間誘導され、4%パラホルムアルデヒドで固定されました。次に、分化した骨芽細胞と脂肪細胞をアリザリンレッド染色とオイルレッドO染色(武漢Pulande Biological Technology Co.、Ltd。、中国武漢)で実施し、顕微鏡で観察しました。
エクソソームの分離と同定
4継代のBMSCを48時間培養して上清を回収し、遠心分離(800gおよび2000g)し、0.22μmおよび100,000 MWフィルターメンブレンでろ過し、遠心分離(100,000 g)して沈殿物を回収しました。次に、沈殿物をPBSで再懸濁し、100,00gで再度遠心分離してエクソソーム沈殿物を得た。 PBS中のBMSC-Exo懸濁液を、ビシンコニン酸(BCA)による濃度検出と、ウエスタンブロット分析(Proteintech、シカゴ、イリノイ州、米国)によるエクソソームメーカータンパク質検出(CD63、CD81、およびCD9)にかけました。
組換えアデノウイルス感染は、BMSCのmiR-133a遺伝子改変を仲介します
BMSCは一晩継代されました。正常対照(等量のPBS)、miR-133a陰性対照(NC)、miR-133a過剰発現(Ad-miR-133a)、およびmiR-133a低発現(Adas-miR-133a)( Hanbio Biotechnology Co.、Ltd.、Shanghai、China)は、100の感染多重度(MOI)に沿ったBMSCでトランスフェクトされました。 BMSCを培養し、対応するエクソソーム(NC Exo 、NC Exo 、Ad-miR-133a Exo 、およびAdas-miR-133a Exo )は超遠心分離によって得られました[20]。
ラットにおけるVMCモデルの確立と実験動物のグループ化
成熟したオスのSPFグレードのSDラットを10のグループに分け、それぞれ8匹のラットを使用しました。コクサッキーウイルスB3(CVB3)は、中国医科学アカデミーの医療バイオテクノロジー研究所(北京、中国)から提供されました。
CVB3(10mg / kg)をラットに腹腔内注射し、一方、PBSまたはBMSC-Exo(100μg)を尾静脈から注射した。対照の正常ラットにCVB3培養液とPBSを注射した。 10 mg / kg CVB3を注射したラットには、さらにPBS、MSC exo を注射しました。 、NC Exo 、Adas-miR-133a Exo 、Ad-miR-133a Exo 、si-NC、またはsi-MAML1(RIBOBIO、広州、中国)。
ラットに7日間連続注射し、眼球の血液を採取した。血液を遠心分離し、血清を収集してサブパッケージ化し、-20°Cで保存しました。ラットを安楽死させた後、心臓標本を採取し、10%ホルムアルデヒドで固定し、勾配アルコールで脱水し、キシレンで透明にし、パラフィンで包埋し、組織学的観察のために切片にした。一部の切片は、分子生物学実験材料として−80°Cに配置されました。
心エコー検査
ウイルス注射後7日目に、ラットにペントバルビタールナトリウム25mg / kgを腹腔内注射した。完全に麻酔した後、電極針に接続された心電図装置の肢リードをラットの肢の端に皮下挿入し、肢リード心電図を記録した。次に、ラットを少し左の仰臥位に固定し、胸部を脱毛し、IIリード心電図を接続して、胸骨傍4腔心臓セクションの大動脈の血流パルスのドップラースペクトルを取得しました。指標には、左心室後壁の厚さ(LVPW)、左心室収縮末期径(LVID)、左心室短縮率(FS)、および左心室駆出率(LVEF)が含まれます。
ヘマトキシリン-エオシン(HE)染色
組織を4%パラホルムアルデヒドで固定し、脱水し、透明にし、パラフィンで包埋しました。次に、4μmの切片を脱ロウし、ヘマトキシリン(Servicebio、武漢、中国)で染色し、1%塩酸アルコールで分化させ、青色に戻し、エオシンで染色し、脱水し、キシレンで透明にし、中性ガムで密封しました。光学顕微鏡(オリンパス、東京、日本)で観察しました。
マッソンコラーゲン染色
パラフィン切片を脱蝋し、2分未満でヘマトキシリンで染色し、立春マゼンタ溶液で染色し、0.5%氷酢酸溶液ですすいだ。次に、切片を1%リン酸アルミニウム水溶液で染色し、暗赤色から明るい赤色、ピンクに染色し、顕微鏡で観察した。次に、切片をアニリンブルー(Pulande)で染色し、通常はキシレンで脱水して密封しました。医療画像分析ソフトウェアImage-Proplus6.0を使用して、コラーゲン線維の陽性染色面積、およびコラーゲン体積分率(CVF)=コラーゲン面積/総視野面積を測定しました。コラーゲン線維の染色位置と色が区別されました(心筋細胞は赤で、コラーゲン線維は細胞間空間で青い縞模様または均質な構造でした)。
末端デオキシヌクレオチドトランスフェラーゼを介したデオキシウリジン三リン酸-ビオチンニックエンドラベリング(TUNEL)染色
パラフィン切片を脱ろうし、クエン酸緩衝液に入れ、350Wで10分間焼いた。切片に50μLのTUNEL溶液を加え、50μLの変換剤-ペルオキシダーゼと結合し、DABで現像し、顕微鏡で観察しました。切片をヘマトキシリンに入れ、95%エタノールI–IIに浸し、無水エタノールI–II、キシレンI–IIと結合し、密封しました。結果は光学顕微鏡で分析されました。
酵素結合免疫吸着測定法(ELISA)
腫瘍壊死因子α(TNF-α)、インターロイキン(IL)-1βおよびIL-6は、ELISAキット(BOSTER Biological Technology Co. Ltd.、Wuhan、China)によって検出されました。眼球血液を604gで遠心分離して上部血清を収集した。細胞培養培地からの遠心分離によって得られた上清は、細胞実験で検出された。サンプルの希釈標準には7つの濃度勾配がありました。ブランクウェルをサンプル希釈液と結合し、別のウェルにテトラメチルベンジジン(TMB)を添加し、各濃度に2つの複製ウェルを設定しました。サンプルウェルを50μLのサンプル希釈液と順番に結合しました。各ウェルを100μLの一次抗体(TMBウェルを除く)と1時間反応させ、300μLの0.01 Mトリス緩衝生理食塩水(TBS)および100μLのアビジン-ビオチン-ペルオキシダーゼ複合体作業溶液(TMBウェルを除く)と反応させました。 )。次に、各ウェルに300μLの0.01 M TBSを添加し、100μLTMBとインキュベートしました。光学密度(OD)値と各ウェルの濃度をすぐに測定し、検量線を作成しました。
逆転写定量的ポリメラーゼ連鎖反応(RT-qPCR)
心筋組織および心筋細胞におけるMiR-133a、コラーゲンΙ、コラーゲンIII、α-SMA、TGF-β1、CTGF、E-カドヘリン、およびFSP-1の発現がRT-qPCRによって検出されました。全RNAを心筋細胞または心筋組織から抽出し、RNA抽出キット(Takara、Dalian、China)を介してcDNAに逆転写し、RT-PCRプライマーをInvitrogen(Guangzhou、China)を介して合成しました。配列を表1に示します。 2 -△△ Ct に従って、ローディングコントロール遺伝子としてグリセルアルデヒド-3-リン酸デヒドロゲナーゼ(GAPDH)またはU6を使用して、相対的な定量的遺伝子発現を分析しました。 メソッド。
<図>ウエスタンブロット分析
ラットを麻酔で安楽死させた。心筋組織を凍結し、液体窒素で粉砕した。次に、プロテアーゼ阻害剤であるフェニルメタンスルホニルフルオリドストック溶液を細胞溶解緩衝液と1:100の比率で混合しました(Beyotime Biotechnology Co.、Ltd.、Shanghai、China)。サンプルを混合溶液で溶解し、細胞のタンパク質を抽出しました。総タンパク質濃度はBCAキットで検出されました。サンプルを5×ローディングバッファーと4:1で混合し、沸騰水浴で10分間実施し、氷浴し、遠心分離しました。電気泳動分離を行い、タンパク質を電気泳動溶液でポリフッ化ビニリデン(Servicebio)メンブレンに転写しました。次に、膜を5%スキムミルクパウダーでブロックし、一次抗体CD63、CD81、およびCD9(Proteintechのウサギ抗ラットポリクローナル抗体、1:100)、MAML1(ab65090、Abcam、MA、USA、1: 1000)、およびGAPDH(Santa Cruz Biotechnology、Inc、Santa Cruz、CA、USA、1:1000)。次に、膜に二次抗体である西洋ワサビペルオキシダーゼ標識IgG(Cell Signaling Technology、Beverly、MA、USA、1:1000)を滴下し、強化された化学発光反応溶液(Pierce、Rockford、IL、USA)に浸しました。ローディングコントロールとしてGAPDHを使用し、ImageJ2xソフトウェアを使用してタンパク質インプリンティング画像を分析しました。
心筋細胞の培養と継代
3〜5日齢のSDラット(浙江大学医学部実験動物センター、浙江、中国)を採取した。心室部分を予冷したハンクス平衡塩類溶液ですすぎ、細かく切り、0.25%トリプシンで分離しました。剥離を停止させるために適切な量の10%完全培地を断片に加え、151gで遠心分離した。 20%FBSを含むDMEMを細胞再懸濁に適用しました。心筋細胞を示差付着法により精製し、トリパンブルー染色により生存率を観察し、生存した心筋細胞を培養した。 24時間後、心筋細胞が壁に付着し、脈動を開始しました。 72時間後、仮足は拡大しました。
心筋細胞のVMCモデルの構築
対数増殖期の4継代の心筋細胞を選択し、MSC Exo に感染させました。 、NC Exo 、Adas-miR-133a Exo 、およびAd-miR-133a Exo 。 100 Tcid50 CVB3ウイルス溶液(100μL)を細胞に添加して、細胞VMCモデルを誘導しました。同時に、コントロール用の細胞に等量の維持液を添加し、47時間培養して1時間感染させた後、対応するエクソソームを心筋細胞に結合させました。
Cell Counting Kit(CCK)-8アッセイ
CCK-8細胞検出キット(Beyotime)を適用して、心筋細胞の生存率を検出しました。対数増殖期に成長したとき、細胞を0.25%トリプシンで剥離し、2.5×10 4 で96ウェル細胞培養プレートに播種しました。 細胞/ウェルあたり。 CCK-8溶液(10μL/ウェル)と結合し、細胞を1〜4時間継続的に培養し、OD 450 nm 値はマイクロプレートリーダーを介して測定されました。
フローサイトメトリー
アネキシンV-APC /ヨウ化プロピジウム(PI)二重染色法を適用して細胞アポトーシスを検出しました。細胞を遠心分離し、250μLの結合バッファー(4mLの結合バッファー+12 mLの脱イオン水)で再懸濁し、1×10 6 に調整しました。 細胞/ mL。 100μLの細胞懸濁液に5μLのアネキシンV-APC(BD Biosciences)と5μLのPI溶液(BD Biosciences)を加え、フローサイトメーターにロードし、コンピューターで自動的に分析しました。
デュアルルシフェラーゼレポーター遺伝子アッセイ
MAML1 3非翻訳領域(UTR)の野生型(wt)または変異型(mut)配列を、pGL3-Mベクター(Promega、WI、USA)にクローン化し、次にMAML1-3-UTR-wtまたはMAML1-にクローン化しました。 3-UTR-mutが生成されました。ベクターは、miR-133aミミックまたはNCとともに、Lipofectamine 2000を介して心筋細胞にコトランスフェクトされました。ルシフェラーゼ活性は、48時間後にデュアルルシフェラーゼレポーター遺伝子システム(Promega)によってテストされました[21]
RNA免疫沈降(RIP)アッセイ
RIPキット(Millipore、USA)を使用して、MAML1とmiR-133aの結合を検出しました。細胞を放射性免疫沈降アッセイバッファー(P0013B、Beyotime、上海、中国)で溶解し、1400 gで遠心分離し、抗体とインキュベートして共沈させました。磁気ビーズ(50μL)を100μLのRIP洗浄バッファーに再懸濁し、5 gの抗MAML1抗体(1 g / mL、ab155786)またはIgG(1:100、ab172730)とインキュベートしました。磁気ビーズ-抗体複合体を900μLのRIP洗浄バッファーに再懸濁し、100μLの細胞抽出物と相互作用させ、プロテイナーゼKで消化し、RT-qPCRで検出しました[22]。
統計分析
データの分析には、SPSS 21.0統計ソフトウェア(IBM Corp. Armonk、NY、USA)を適用しました。測定データは、平均±標準偏差として表されました。 t検定は、2つのグループ間の比較に適用されました。グループ間の比較には一元配置分散分析(ANOVA)を使用し、ペアワイズ比較にはテューキーの事後検定を使用しました。 P で有意であった場合、予測子は保持されました 0.05以下の値。
結果
BMSCとBMSC-Exoの識別
顕微鏡的には、BMSCは紡錘状で丸みを帯びており、渦または放射状のパターンで壁に付着していました(図1A)。脂肪生成誘導のオイルレッドO染色後、4番目のBMSCの脂肪滴は赤くなり、さまざまなサイズの丸い脂肪滴がありました(図1B)。骨形成の誘導後、石灰化結節を発現する細胞は、アリザリンレッド染色後に赤色を示し、石灰化結節および重複細胞の不均一な分布を示した(図1C)。フローサイトメトリーは、MSCマーカーCD29およびCD44(> 95%)が発現していることを示しましたが、造血幹細胞表面抗原CD34(<95%)は発現していませんでした(図1D)。これらの結果は、BMSCが高純度であり、International Society of CellTherapyのMSC基準に準拠していることを示しています。
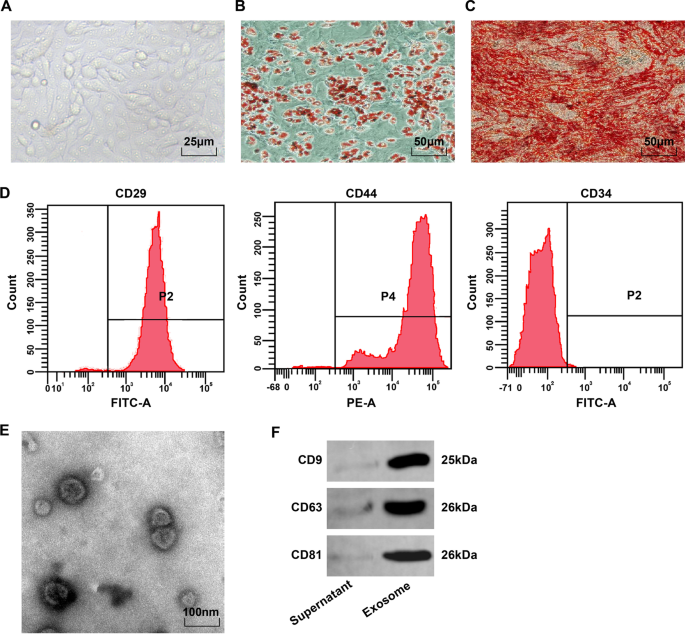
BMSC表現型の観察とBMSC-Exoの同定。 A 4継代におけるBMSCの形態学的観察; B 脂肪細胞のオイルレッドO染色の結果。 C 骨芽細胞のアリザリンレッド染色の結果; D フローサイトメトリーで検出されたBMSCの表現型。 E BMSC-Exoの電子顕微鏡観察; F CD9、CD63、およびCD81のタンパク質バンド
透過型電子顕微鏡は、BMSC-Exosが明確な周辺膜構造、異なるサイズ、および40〜100 nmの直径を持つ楕円形の小胞であることを観察しました(図1E)。ウエスタンブロット分析は、抽出された産物がCD9、CD63、およびCD81のエクソソーム由来の特徴的なタンパク質を発現することを示しました(図1F)。
エクソソームmiR-133aの上昇は心筋炎の症状を改善します
miR-133a組換えアデノウイルスによるBMSCのトランスフェクションが観察されました(図2A)。 NC、Ad-miR-133a、およびAdas-miR-133aの多数の緑色蛍光発現が倒立蛍光顕微鏡下で観察され、組換えアデノウイルスベクターがBMSCを効果的にトランスフェクトできることを示しています。 miR-133aのトランスフェクション効率をテストするために、BMSCおよびそれらのエクソソームにおけるmiR-133aの発現をRT-qPCRで測定しました。 miR-133aのアップレギュレーションはmiR-133aの発現を増加させ、miR-133aのダウンレギュレーションはmiR-133aの発現を減少させることが発見されました(図2B)。続いて、miR-133aを含むエクソソームをラットに注射しました。倒立蛍光顕微鏡下で、NC Exo の処理後にVMCラットで緑色蛍光発現が観察されました。 、Ad-miR-133a Exo 、またはAdas-miR-133a Exo 、組換えアデノウイルスベクターがラットの心筋組織に感染したことを示しています(図2C)。 RT-qPCR実験では、VMCラットでのmiR-133aの発現が明らかに減少していることもわかりました。 miR-133aの発現は、Ad-miR-133a Exo を注射したVMCラットで明らかに上昇しました。 しかし、Adas-miR-133a Exo を注射したVMCラットでは減少しました (図2D)。ラットの全身状態については、正常対照ラットの全身状態は正常であり、VMCの特徴はAdas-miR-133a Exo > 、ざらざらした無秩序な髪、呼吸困難、および少しの食事療法など。 MSC Exo で治療されたVMCラット 、NC Exo 、およびAd-miR-133a Exo 、これらの兆候はさまざまな程度に改善されました。 VMCラットの体重は、感染後1日から継続的に減少し、MSC Exo を注射しました。 、NC Exo 、またはAd-miR-133a Exo ラットの体重が増加した。 Ad-miR-133a Exo で治療されたVMCラットの体重 明らかに増加し、Adas-miR-133a Exo を注射したVMCラットの体重が増加しました 明らかに減少しました(図2E)。
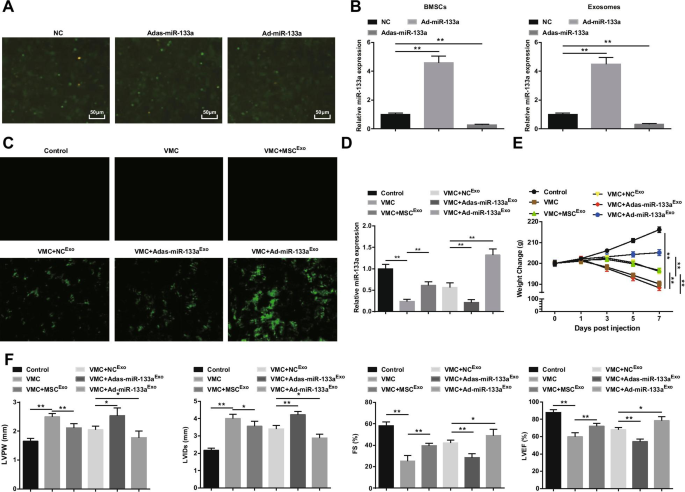
アップレギュレーションされたエクソソームmiR-133aは心筋炎を軽減します。 A miR-133a組換えアデノウイルスのBMSCトランスフェクション。 B miR-133aを調節した後のBMSCおよびそれらのエクソソームにおけるmiR-133a発現のRT-qPCR検出。 C miR-133aトランスフェクション効率は倒立蛍光顕微鏡でテストされています。 D RT-qPCRを介してテストされた心筋組織におけるmiR-133aの相対的発現。 E 各グループのラットの体重変化; F 各グループのラットにおけるLVPW、LVID、FSおよびLVEFの決定。 * P <0.05; ** P <0.001
心筋機能の観察は(図2F)、VMCラットがLVPWおよびLVIDを増加させ、FSおよびLVEFを減少させたことを示唆した。エクソソーム注射後、LVPWとLVIDが低下し、VMCラットで明らかに上昇したFSとLVEFが見られました。 Adas-miR-133a Exo Ad-miR-133a Exo の治療が損なわれている VMCラットの心筋機能の改善。
アップレギュレーションされたエクソソームmiR-133aは、VMCラットの心筋組織の炎症を抑制します
HE染色は、正常な対照ラットの心筋線維が密接に配置されており、間葉に炎症性細胞浸潤がなかったことを示した。 VMCラットの心筋細胞は無秩序であり、間葉は多数の炎症細胞によって浸潤されていた。 MSC Exo を注射したVMCラットの心筋細胞 またはNC Exo 間葉に少量の炎症細胞が浸潤して、整然と配置されていました。 Adas-miR-133a Exo 後のVMCラットの心筋細胞 治療は無秩序に行われ、間葉の炎症細胞が浸潤していた。 Ad-miR-133a Exo で処理されたVMCラットの心筋細胞 明らかな炎症細胞の浸潤なしに整然と配置されました(図3A)。
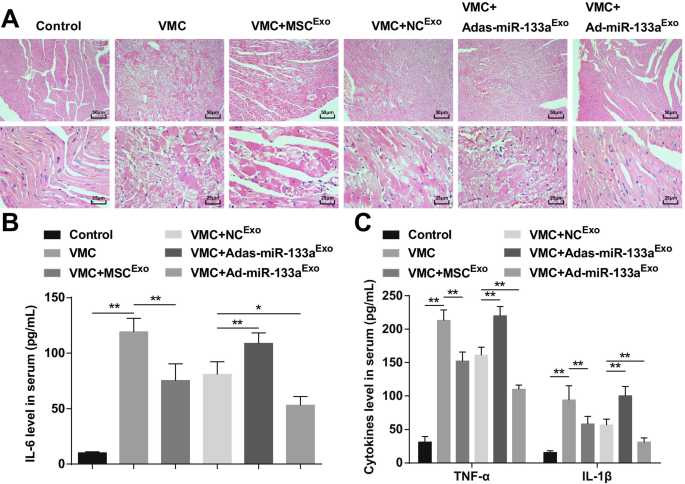
エクソソームmiR-133aの増加は、VMCを伴う心筋組織の炎症を抑制します。 A 各グループのラット心筋組織のHE染色。 B ELISAを介して試験された血清中のIL-6の発現; C ELISAを介して試験された血清中のTNF-αおよびIL-1βの発現。 * P <0.05; ** P <0.001
ELISAは、(図3B、C)炎症性因子(TNF-α、IL-1β、およびIL-6)がVMCラットで明らかに増加したことを示した。 Ad-miR-133a Exo を注射したVMCラット 炎症性因子のレベルが低下していました。 Adas-miR-133a Exo 治療はVMCラットの炎症性因子の上昇を引き起こしました。
上昇したエクソソームmiR-133aは、VMCのラットの心筋組織のCVFを低下させます
マッソン染色は、正常なラットの心筋線維が密接に配置されており、青いコラーゲン線維がほとんどないことを明らかにした。 CVB3注射後、心筋細胞は肥大型であり、結合組織の過形成と多数の青いコラーゲン線維を伴い、CVFは明らかに上昇していました。エクソソームで処理すると、心筋細胞が整然と配置され、細胞間結合組織の過形成が減少し、青いコラーゲン線維とCVFが明らかに減少しました。 Adas-miR-133a Exo を注射したVMCラットの心筋細胞間空間 広がり、細胞は明らかに拡大し、青いコラーゲン繊維とCVFは明らかに増加しました。 Ad-miR-133a Exo を使用したVMCラットでは、細胞間スペースが減少し、青いコラーゲン線維の分布とCVFが減少しました。 治療(図4A、B)。
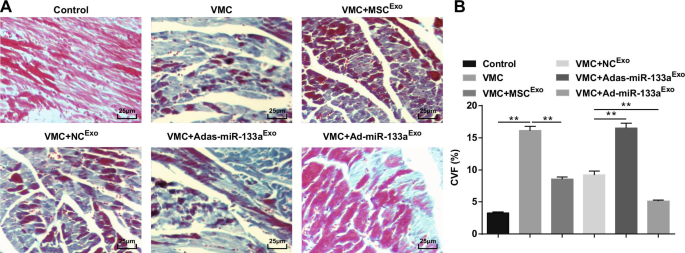
アップレギュレーションされたエクソソームmiR-133aは、VMCのラットの心筋組織のCVFを低下させます。 A ラットの心筋組織のマッソン染色; B 各グループのラットのコラーゲン体積分率。 ** P <0.001
増加したエクソソームmiR-133aは、VMCを有するラットの心筋組織におけるコラーゲンI、コラーゲンIII、TGF-β1、およびCTGFの発現を低下させます
コラーゲンIとコラーゲンIIIはコラーゲンの主成分であり、主に細胞結合や細胞膜、細胞間物質、細胞質に分布しています。 TGF-β1とCTGFは線維症の特徴的なタンパク質です。 RT-qPCRの結果は、コラーゲンI、コラーゲンIII、TGF-β1、およびCTGF mRNAの発現レベルがVMCラットで増加したが、エクソソーム処理後に減少したことを示しました。 Ad-miR-133a Exo で治療されたVMCラット Adas-miR-133a Exo 後のVMCラットでは、コラーゲンI、コラーゲンIII、TGF-β1、CTGFのmRNA発現レベルが低下していました。 治療は反対の状況を示しました(図5A–C)。
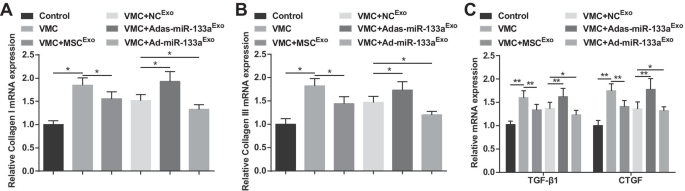
Elevated exosomal miR-133a reduces the mRNA expression of collagen I, collagen III, TGF-β1 and CTGF in myocardial tissues of rats with VMC. A Collagen I mRNA expression in myocardial tissues of rats was detected by RT-qPCR; B Collagen III mRNA expression in myocardial tissues of rats was detected by RT-qPCR; C TGF-β1 and CTGF mRNA expression in myocardial tissues of rats was detected by RT-qPCR. * P <0.05; ** P <0.001
Up-regulated Exosomal miR-133a Inhibits the Cardiomyocyte Apoptosis in Myocardial Tissues of Rats with VMC
TUNEL staining showed that the apoptotic cardiomyocytes were brownish black or brownish yellow with nuclear condensation. The number of apoptotic cells was increased in VMC rats which would be attenuated by exosome treatment. The VMC rats injected with Ad-miR-133a Exo had reduced number of apoptotic cells and those injected with Adas-miR-133a Exo had increased number of apoptotic cells (Fig. 6A, B).

Increased exosomal miR-133a inhibits the cardiomyocyte apoptosis in myocardial tissues of rats with VMC. A TUNEL staining of rat myocardial tissues in each group; B The number of TUNEL positive cells in each group. ** P <0.001
Elevated Exosomal miR-133a Depresses EMT in Myocardial Tissues of Rats with VMC
E-cadherin, α-SMA, and FSP-1 are key indicators of EMT. Results of RT-qPCR demonstrated that α-SMA and FSP-1 mRNA expression levels were elevated and E-cadherin mRNA expression level was decreased in VMC rats. In addition, α-SMA and FSP-1 mRNA expression levels were reduced and E-cadherin mRNA expression level was increased in VMC rats after exosome treatment. α-SMA and FSP-1 mRNA expression levels were elevated and E-cadherin mRNA expression level was decreased in VMC rats treated with Adas-miR-133a Exo , while the expression of these indicators was opposite in VMC rats injected with Ad-miR-133a Exo (Fig. 7A, B).

Up-regulated exosomal miR-133a represses EMT in myocardial tissues of rats with VMC. A The mRNA expression of α-SMA in rat myocardial tissues in each group was detected by RT-qPCR; B The mRNA expression of FSP-1 and E-cadherin in rat myocardial tissues in each group was detected by RT-qPCR. * P <0.05; ** P <0.001
Up-regulated Exosomal miR-133a Depresses Inflammation of Cardiomyocytes in VMC
As a result, fluorescence microscopy captured green fluorescent expression in VMC rats treated with NC Exo , Ad-miR-133a Exo , or Adas-miR-133a Exo , indicating that the recombinant adenovirus vector infected cardiomyocytes of rats (Fig. 8A). RT-qPCR and ELISA discovered that (Fig. 8B, D) miR-133a expression was reduced and inflammatory factors (TNF-α, IL-1β, and IL-6) were increased in VMC rats, which would be reversed by exosome treatment. The VMC rats treated with Ad-miR-133a Exo had up-regulated miR-133a and decreased inflammatory factors in VMC rats, while those treated with Adas-miR-133a Exo presented decreased miR-133a and increased levels of inflammatory factors in VMC rats.

Elevated exosomal miR-133a restrains inflammation of cardiomyocytes in VMC. A miR-133a transfection efficiency tested via inverted fluorescence microscope; B The relative expression of miR-133a in cardiomyocytes of rats in each group; C IL-6 expression in culture supernatant of cardiomyocytes in each group; D TNF-α and IL-1β expression in culture supernatant of cardiomyocytes in each group. * P <0.05; ** P <0.001
Elevated Exosomal miR-133a Promotes Cell Viability, and Represses Apoptosis of Cardiomyocytes in VMC
The apoptosis and the cell viability were detected via AnnexinV-APC/PI double staining and CCK-8 assay. The results revealed that there was an obvious increase in apoptosis rate, a decrease in cell viability of cardiomyocytes in VMC rats. Exosome treatment reduced apoptosis rate and enhanced the viability of cardiomyocytes. Adas-miR-133a Exo enhanced the apoptosis rate and disrupted the viability of cardiomyocytes in VMC rats. Ad-miR-133a Exo treatment functioned the opposite effects on cardiomyocytes of VMC rats (Fig. 9A–C).

Increased exosomal miR-133a promotes viability and represses apoptosis in cardiomyocytes in VMC. A The cardiomyocytes apoptosis detected via flow cytometry; B Quantification results of A; C The cell viability detected via CCK-8 assay. * P <0.05; ** P <0.001
miR-133a Targets MAML1
It has been reported that up-regulated miRNA-193b reduces myocardial I/R damage by targeting MAML1 [18]. Based on that, we cross-screened downstream genes of miR-133a through bioinformatics websites PITA, miRanda, PicTar, microT and miRmap, and selected MAML1 as a target of miR-133a (Fig. 10A). We constructed MAML1-wt or MAML1-mut, and co-transfected cardiomyocytes with miR-133a mimic or NC. The results showed that miR-133a mimic reduced the luciferase activity of MAML1-wt (Fig. 10B). The RIP experiment further verified the targeting relationship between miR-133a and MAML1 (Fig. 10C). RT-qPCR and Western blot detection of MAML1 expression showed that MAML1 expression was decreased in cardiomyocytes transfected with miR-133a mimic (Fig. 10D, E).

miR-133a targets MAML1. A miR-133a’s targets predicted on bioinformatics websites; B The targeting relationship between miR-133a and MAML1 verified by dual luciferase reporter gene experiment; C The targeting relationship between miR-133a and MAML1 verified by RIP experiment; D /E MAML1 expression changes after up-regulation of miR-133a detected by RT-qPCR and Western blot. * P <0.05; ** P <0.01; *** P <0.001
Inhibition of MAML1 has a Protective Effect on Rats with Myocarditis and Reverses the Effect of miR-133a-Inhibited Exosomes on Rats with VMC
To further study the effect of miR-133a-regulated MAML1 on rats with VMC, we injected si-MAML1 or si-NC adenovirus into VMC rats or VMC rats that had been treated with miR-133a-silenced exosomes. The injection success was validated by RT-qPCR and Western blot (Fig. 11A, B). The results manifested that injection of si-MAML1, the weight of VMC rats was increased (Fig. 11C), cardiac function was improved (Fig. 11D–G), myocardial tissue pathology and fibrosis were attenuated (Fig. 12A–C), serum inflammation (Fig. 12D, E) and cardiomyocyte apoptosis (Fig. 13A–G) were inhibited. Also, the deleterious effects of miR-133a-silenced exosomes in VMC rats were reversed after injection of si-MAML1.

Inhibition of MAML1 has a protective effect on myocarditis rats and can reverse the effect of miR-133a-silenced exosomes on rats with VMC. A /B MAML1 expression in myocardial tissue of rats detected by RT-qPCR and Western blot; C. Weight change of rats; D –G Determination of LVPW, LVIDs, FS and LVEF in rats; ** P <0.01; *** P <0.001; ****P <0.0001

Inhibition of MAML1 can reverse the effect of miR-133a-silenced exosomes on rats with VMC. A HE staining of rat myocardial tissue; B Masson staining of myocardial tissues in rats; C CVF of rats; D The expression of IL-6 in serum tested via ELISA; E The expression of TNF-α and IL-1β in serum tested via ELISA.****P <0.0001

Inhibition of MAML1 can reverse the effect of miR-133a-inhibiting exosomes on rats with VMC. A Collagen I mRNA expression in myocardial tissues of rats was detected by RT-qPCR; B Collagen III mRNA expression in myocardial tissues of rats was detected by RT-qPCR; C TGF-β1 and CTGF mRNA expression in myocardial tissues of rats was detected by RT-qPCR; D TUNEL staining of rat myocardial tissues in each group; E The number of TUNEL positive cells in each group. F The mRNA expression of α-SMA in rat myocardial tissues in each group was detected by RT-qPCR; G The mRNA expression of FSP-1 and E-cadherin in rat myocardial tissues in each group was detected by RT-qPCR. * P <0.05; ** P <0.01; ****P <0.0001
Discussion
Myocarditis is an inflammatory heart illness resulting in DCM and heart failure and is most frequently induced by viral infections such as CVB3 [2]. A study has revealed that miR-133 relieves cardiomyocyte apoptosis and electrical remodeling in mice with VMC [23]. Additionally, changed exosomal miRNAs are also found to be linked with the pathogenesis of CVB3-induced myocarditis [14]. Exosomes derived from cardiac progenitor cells ease CVB3-induced apoptosis via restraining the proliferation of CVB3 in VMC [24]. This study explored the regulatory mechanism of BMSC-derived exosomal miR-133 on myocardial fibrosis and EMT in VMC rats (Additional file 1:Fig. 1).
The study found that the expression of miR-133a was decreased in VMC. As demonstrated before, miR-133a expression is decreased in MI [7]. A study has also suggested that the relative expression of miR-133 in mouse hearts of the VMC is obviously decreased with contrast to the controls [23]. There are some connections of miRNAs with exosomes. The differential expression of exosomes and of exosomal miRNAs in illness has been regarded as biomarkers of disease with performance of noninvasive clinical diagnosis together with their therapeutic potentials [25]. Lin et al. have found that miR-133 is specially sorted into hypoxia/reoxygenation (H/R)-caused human endothelial progenitor cells-derived exosomes to increase fibroblast angiogenesis and EMT [26]. Another study has revealed that MSCs exhibits a communication with brain parenchymal cells and may modulate neurite outgrowth by transfer of miR-133b to neural cells via exosomes [27].
The major finding of this work manifested that up-regulated exosomal miR-133a promoted cell viability, inhibited inflammation, apoptosis, EMT, and fibrosis in rats with VMC. They suits well with a former research that miR-133a silence reverses the Astragalus polysaccharides treatment-induced osteosarcoma MG63 cell proliferation inhibition, together with cell apoptosis promotion [28]. Another study has revealed that overexpressed miR-133a suppresses angiogenesis, apoptosis, fibrosis, and inflammation, while accelerating therapeutic cardiac remodeling in ischemic myocardial illnesses [29]. Similar to our study, Li et al. have stated that miR-133 inhibits cardiomyocyte apoptosis by regulating the expression of apoptosis-related genes in the hearts of VMC mice [23]. The over-expressed miR-133a has been reported to depress hypoxia-induced apoptosis and strengthen cardiomyocyte survival [30]. Meanwhile, the up-regulated serum exosomal miR-30a and miR-181d may have the potentials to be applied as biomarkers for VMC diagnosis [14].
Another finding in our study was that up-regulated exosomal miR-133a decreased CVF, reduced the expression of collagen I and collagen III in rats with VMC. A article has elucidated that released fibroblast growth factor-18 from a collagen membrane causes osteoblastic activity participating in down-regulated miR-133a [31]. In vitro excessive expression of miR-133a depresses cardiomyocyte hypertrophy and reduces collagen expression [32], as evidenced in another study. CVF equals the ratio of collagen area to the sum of myocardial area and collagen area, and the mean value shows the CVF of the section [33]. This finding is also reported by Wang et al. that VMC mice model is successfully constructed by CVB3 infection, manifesting apparent higher CVF expression in contrast with the control group [34]. Moreover, the finding is consistent with that of Ferreira et al. who demonstrates that miR-133a may take on a major role in the modulation of gene expression in chronic Chagas disease cardiomyopathy pathogenesis, with potential link as diagnostic and prognostic tools [8]. Furthermore, evidence has shown that knocking down MAML1 can reduce the hypertrophy of pre-treated cardiomyocytes [35]. In our study, we found that MAML1 was the target gene of miR-133a and inhibition of MAML1 reversed the effects of miR-133a-silenced exosomes on rats with VMC. In myocardial ischemia–reperfusion injury, miR-193b-mediated down-regulation of MAML1 could in part reduce infarction and myocardial enzymes, as well as attenuate apoptosis of cardiomyocytes [18]. Also, there is a report suggesting that deficiency of MAML1 could relieve hepatic fibrogenesis [19].
Conclusion
In conclusion, this present study offers evidence that miR-133a is down-regulated in rats with VMC, and elevated exosomal miR-133a improves cardiac function and restrains myocardial fibrosis and EMT in rats with VMC, as well as enhances viability and represses apoptosis of cardiomyocytes in VMC through targeting MAML1. Our study also suggests that inhibition of MAML1 has a protective effect on rats with myocarditis and reverses the effect of miR-133a-inhibited exosomes on rats with VMC. The identification of the exosomal miR-133a in myocardial fibrosis and EMT of myocarditis may potentially widen our understanding of mechanisms underpinning myocarditis and also bear clinical value as a novel molecular target. More researches should be undertaken for making inroads into the treatment of this disease.
略語
- miR-133:
-
MicroRNA-133
- BMSC-Exo:
-
Bone marrow mesenchymal stem cell-derived exosome
- EMT:
-
Epithelial–mesenchymal transition
- VMC:
-
Viral myocarditis
- CVF:
-
Collagen volume fraction
- DCM:
-
Dilated cardiomyopathy
- CVB3:
-
Coxsackie B3 virus
- miRNAs:
-
MicroRNAs
- MI:
-
Myocardial infarction
- SPF:
-
Specific pathogen-free
- SD:
-
Sprague–Dawley
- DMEM:
-
Dulbecco’s Modified Eagle Medium
- PBS:
-
リン酸緩衝生理食塩水
- FBS:
-
ウシ胎児血清
- NC:
-
Negative control
- MOI:
-
Multiplicity of infection
- LVPW:
-
Left ventricular posterior wall thickness
- LVIDs:
-
Left ventricular end-systolic diameter
- LVEF:
-
Left ventricular ejection fraction
- HE:
-
Hematoxylin–eosin
- TUNEL:
-
Terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate-biotin nick end-labelin
- ELISA:
-
酵素免疫測定法
- TMB:
-
Tetramethylbenzidine
- TBS:
-
Tris-buffered saline
- ABC:
-
Avidin–Biotin-Peroxidase Complex
- OD:
-
光学密度
- RT-qPCR:
-
Reverse transcription quantitative polymerase chain reaction
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- CTGF:
-
Connective tissue growth factor
- CCK:
-
Cell counting kit
- PI:
-
ヨウ化プロピジウム
- ANOVA:
-
Analysis of variance
ナノマテリアル
- 黑リンナノ粒子は、TG2発現のアップレギュレーションを通じてEMSCの骨形成分化を促進します
- 遷移金属をドープしたカオリナイトナノクレイの構造と電子特性
- 自己組織化と急速熱アニーリングによるAgナノヘアによる「厚く生い茂った」活性炭繊維
- 原子間力顕微鏡によるポリスチレン薄膜の接着力とガラス転移の研究
- 二層/三層ブロードバンドSiO2反射防止膜の断面形態に関するTEMおよびSTEM研究
- ラットにおける腹腔内および静脈内投与経路による生合成された銅および酸化亜鉛ナノ粒子の比較invivo精査
- 生体ヒドロキシアパタイトに基づくSr含有ガラスセラミック複合材料の開発と特性評価
- 骨組織再生におけるグラフェンファミリー材料:展望と課題
- 軟骨形成的に分化しているヒト脂肪由来幹細胞の原子間力顕微鏡ベースのナノスコピー:ナノ構造とインテグリンβ1発現
- プルシアンブルーナノ粒子標識間葉系幹細胞:細胞生存率、増殖、遊走、分化、細胞骨格、およびタンパク質発現のinvitroでの評価
- 遷移元素V、Cr、およびMnによってエッジ修飾されたアームチェアブラックフォスフォレンナノリボンの電子特性