


発光分光法による金属分析
発光分光法による金属分析
発光分光技術は、1800年代半ばに実施された実験に端を発していますが、元素分析を実施するための最も有用で柔軟な手段のいくつかであり続けています。自由原子は、エネルギーの高い環境に置かれると、一連の狭い波長間隔で発光します。輝線と呼ばれるこれらの間隔は、輝スペクトルと呼ばれるパターンを形成します。これは、輝線を生成する原子の特性です。線の強度は通常、線を生成する原子の数に比例します。サンプル中の元素の存在は、その特性線の1つまたは複数の励起源からの光の存在によって示されます。この元素の濃度は、線の強度を測定することで決定できます。したがって、特徴的な発光スペクトルは定性的な元素分析の基礎を形成し、輝線の強度の測定は定量的な元素分析の基礎を形成します。鉄とリチウムの発光スペクトルを図1に示します。

図1鉄とリチウムの発光スペクトル
発光分光計としても知られる発光分光器は、通常、(i)さまざまなサンプルタイプの主成分および微量元素成分の定量分析、および(ii)定性元素分析に使用されます。適用例には、(i)鋼およびその他の合金中の合金元素の濃度の迅速な測定、(ii)地質材料の元素分析、(iii)半導体材料中の微量不純物濃度の測定、(iv)摩耗金属の分析が含まれます。油、(v)水性サンプル中のアルカリおよびアルカリ土類濃度の測定、および(vi)セメント中のカルシウムの測定。
サンプルは、導電性の固体(アーク、スパーク、グロー放電)、粉末(アーク)、および溶液(炎)の形をしています。サンプルサイズは、約0.000001グラムから数グラムまでさまざまな特定の手法によって異なります。サンプルの準備は、機械加工または粉砕(金属)、溶解(炎の場合)、および分解または灰化(有機サンプル)によって行われます。
発光分光技術の限界は、(i)窒素、酸素、水素、ハロゲン、希ガスなどの一部の元素を決定することが困難または不可能である、(ii)サンプルの形態が特定の技術と互換性があること、および(iii )すべてのメソッドは、マトリックスに依存する応答を提供します。推定分析時間は、サンプル準備の要件に応じて、30秒から数時間の範囲です。
関連する技術の機能には、(i)X線蛍光はバルクおよび微量成分元素分析用であり、定量分析には高度なデータ削減が必要であり、軽元素(原子番号9)には役立ちません。(ii)誘導結合プラズマ(ICP)発光分光法は、10億分の1の検出限界で、サンプルが溶液中であり、水素、窒素、酸素、ハロゲン化物、および貴ガスには有用ではない、迅速な定量的元素分析用です。(iii)直流プラズマ発光分光法も同様です。 ICP発光分光法の性能において、および(iv)原子吸光分光法は単一チャネル技術であり、多元素分析には非効率的ですが、ほとんどの元素に対して好ましい感度と精度を備えています。
広い意味で、発光分光法には、励起源としてICPを使用するICP発光分光法が含まれます。ただし、「発光分光法」および「発光分光法」という用語は、通常、励起放電を生成するために火花放電、直流アーク放電、グロー放電、または火炎源を使用する発光分光法を指します。この記事では、鉄鋼業界で使用されているため、火花放電を使用した光学分光法について説明します。
多くの発光分光器は、測定の再現性(精度)を向上させるために「パルス分布分析」(PDA)を備えています。この方法は、アルゴン雰囲気中の火花放電から得られた火花パルス生成発光スペクトルの統計的処理を含みます。発光分光器は、固体金属サンプルの迅速な元素分析を提供し、製鋼プロセスの品質管理に不可欠です
一般原則
原子が生成する特徴的なスペクトルは、原子の電子構造を反映しています。原子価または外殻電子のエネルギーの変化により、発光分光法で使用される原子線が生じます。各原子には基底状態があり、そのすべての電子が最小の位置エネルギーの位置を占めています。原子がエネルギーを吸収すると、1つまたは複数の外部電子がより高いエネルギーに昇格し、励起状態が生成されます。原子状態のエネルギーは、個々の電子のエネルギーと、電子間の相互作用から生じるエネルギー変化の関数です。電子配置の可能な組み合わせごとに、原子の状態を表す分光項が生成されます。図2にリチウム原子を例にとった発光スペクトルの原理を示します。
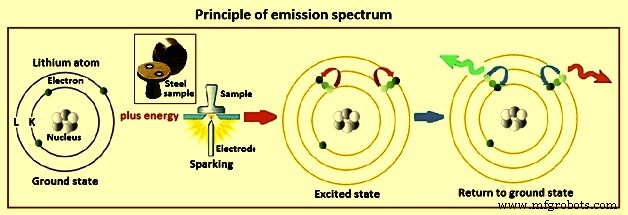
図2発光スペクトルの原理
電子エネルギーレベル –水素やアルカリ金属などの最も単純な原子は、満たされたシェルの外側に1つの電子しかありません。これらの原子の単純な電子配置は、いくつかの可能な項を生成します。原子の輝線は、原子がある励起状態から別の低エネルギー状態に自発的に遷移するときに発生します。可能な状態のすべての組み合わせが輝線を生成するわけではありません。量子機械的に導出された選択規則に従う遷移のみが自発的に発生します。さまざまな要因が線の相対強度を制御します。共鳴遷移と呼ばれる、低励起状態と基底状態の間のこれらの遷移は、一般に最も強い放出をもたらします。
励起された電子のエネルギーは、イオン化限界に達するまで、励起状態間の間隔が狭くなるにつれて増加します。この時点で、電子は原子に結合されなくなり、連続した範囲のエネルギーをとることができます。このような束縛されていない電子は、束縛状態に遷移する可能性があります。遷移の上位状態は離散値に限定されないため、このような遷移からの光は、ある範囲の波長にわたって連続的に拡散します。
原子のイオン化限界は、一価イオンの基底状態に対応します。残りの束縛された電子を励起すると、新しい項系と新しい線のセットが生成されます。イオン化と励起は、原子から電子が完全に取り除かれるまで続く可能性があります。実際の放出源では、イオン化が2つの電子の除去を超えて進行することはめったになく、ほとんどの場合、イオン化の最初の段階のみを考慮する必要があります。ただし、分析では通常、中性原子線の代わりに最初のイオンスペクトルの線が使用されます。
スペクトルのオーバーラップ –元素分析に原子発光を使用するには、サンプル内の他の種からの重複する発光とは関係なく、対象のラインからの発光強度を測定できる必要があります。望ましくないオーバーラップの確率は、スペクトル内のラインの数と、各遷移の波長の広がりまたはライン幅によって異なります。すべての原子項システムが図2のリチウムについて示されているものと同じくらい単純である場合、スペクトルの重複の可能性は低くなります。ただし、リチウムは最も単純な原子の1つです。
より複雑な電子構造を持つ原子は、それに対応して複雑な発光スペクトルを生成します。鉄のスペクトル(図1に示されている)は、そのようなスペクトルの複雑さを示しています。単一元素の1つのイオン化段階からのスペクトルは、十分な励起エネルギーが与えられると、数百の輝線で構成されます。サンプルに複数の元素が存在し、それぞれが中性およびイオンスペクトルを生成する場合、複雑さが増します。
線の広がり –実際には、各輝線が厳密に単色であり、機器が無限のスペクトル分解能で利用できる場合、スペクトルの複雑さは問題になりません。電子用語に関連するエネルギーは正確に定義されていませんが、値の範囲に広がっています。エネルギー準位の不確実性は、輝線の波長の広がりとして輝スペクトルに現れます。いくつかの要因がエネルギーの広がりの大きさを決定します。発光分光法で最も重要なのは、励起源内の他の種との発光原子またはイオンの頻繁な衝突と、不均一な電場へのエミッターの配置です。
最初のタイプのラインブロードニングは衝突ブロードニングであり、2番目のタイプはスタークブロードニングです。 3番目のタイプであるドップラー広がりは、放出を検出するデバイスに対する放出種の動きに起因します。固定遷移エネルギーの場合、検出器に向かって移動する原子から記録された放出は、静止している原子から記録された放出よりも短い波長になります。検出器から離れる方向に移動する原子からの放出は、より長い波長にあります。これらの3つの線を広げる寄与の相対的な大きさは、放出を刺激する発生源のタイプに強く依存します。線幅への衝突の寄与は、主にソース圧力の関数です。特定の要素のドップラー寄与は、ソース温度に依存します。スタークの寄与の大きさは、エミッターの近くの荷電種の密度に依存します。
自己吸収 –上記の効果のいずれかによって生成された原子線プロファイルは、自己吸収によって変更できます。分光源に高濃度の原子がある場合、ある原子が放出する放射線が同じタイプの別の原子に吸収される確率は妥当です。吸収の確率は、翼の近くの波長よりもラインプロファイルの中心に近い波長の方が高くなります。このような条件下で観察された放出プロファイルは、自己吸収がない場合に観察されたものよりも平坦で幅が広い。吸収原子が放出原子よりも低い温度にある場合、線プロファイルは図3に示すものと同様になります。低温吸収体のドップラー吸収プロファイルは、より高温のエミッターの放出プロファイルよりも狭くなります。これは自己逆転と呼ばれます。
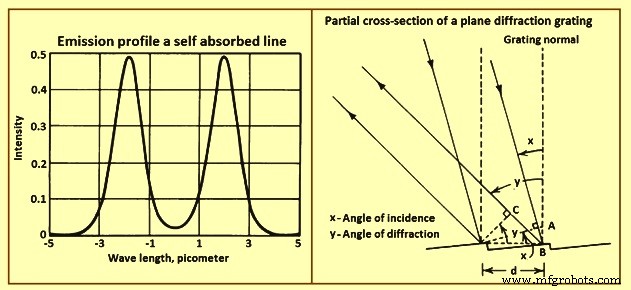
図3自己吸収線の放射プロファイルと平面回折格子の部分断面
分子放出 –分光源のエネルギー放出ボリュームには、遊離原子に加えて小分子が含まれている可能性があります。原子と同様に、分子は、分子の外部電子のエネルギーの変化を反映する発光を生成します。原子とは異なり、分子には各電子状態に関連する多数の振動および回転レベルがあります。分子内の各電子遷移は、遷移に関与する電子状態の振動および回転構造を反映する個々の線で構成される発光バンドを生成します。
分子バンドは、記録されたスペクトルに強いエッジとして現れ、その中から、エッジからの距離とともに増加する間隔で、より高いまたはより低い波長でより弱い線が発達します。エッジはバンドヘッドです。多くの近接した線で構成されているため、分子バンドがスペクトルの領域を支配する可能性があり、その領域の他の種からの発光の検出が複雑になります。放出源は、分子放出を最小限に抑えるように設計されることがよくあります。それほど頻繁ではありませんが、濃度を測定するために原子線強度の代わりにバンド強度が使用されます。
光学システム
原子放出は、1つの原子種からの放出を測定し、他のソースからの放出とは無関係にその強度を記録できる範囲でのみ分析的に有用です。この検出と定量化には、高分解能の波長選別機器が必要です。さらに、光を分類する前に、効率的に収集する必要があります。空間的に不均一な放射源の孤立した領域からのみ収集することもあります。
波長選別装置 –最新の波長選別装置の重要な要素は、回折格子です。これは、多くの近接した平行な溝を持つ正確に成形された反射面です。回折格子の部分断面図を図3に示します。平行光線が回折格子の隣接する溝に当たります。入射光線は互いに同相です。グレーティングから散乱された光線は、さまざまな経路を通過しました。パスの長さの違いはAB+BCです。
波長の整数倍である経路差を生成する角度では、出る光線は同相であり、光はその角度で回折されます。他の角度では、出る光線の位相がずれており、破壊的な干渉が発生します。与えられた波長に対して回折が起こる角度は、AB = dであることに注意することで決定できます。 sinxおよびBC= d sin y where d は回折格子の溝の間隔、xは入射角、yは回折角です。回折条件は、方程式m.lambda =d。(sin x +/- sin y)で与えられます。入射ビームと回折ビームがグレーティング法線の反対側にある場合、マイナス記号が入ります。
発光分光法には、通常、2種類の波長選別装置(図4)が使用されます。最初のグレーティングモノクロメータは、放射線のシングルチャネル検出に使用されます。図4は、典型的な構成であるCzerny-Turnerモノクロメータを通る光路を示しています。光は入口スリットを通ってモノクロメータに入り、コリメートミラーを通過します。コリメートされた光は平面回折格子に当たり、その波長に依存する角度で回折されます。一部の光は、集束ミラーに当たるような角度で回折されます。次に、焦点を合わせて、モノクロメータの焦点面に入口スリット画像のアレイを形成します。スリット画像のアレイ内の位置は、それを形成する光がグレーティングを出た角度によって異なります。出口スリットを中心とする画像の波長は、式mで与えられます。 lambda =2d.sin q.cos pここで、qは格子が回転する角度、pは計器の角度であり、格子の中心といずれかのミラーの中心を通る線が中心線となす角度です。楽器の。 qとpの関係、および最初の式で使用される角度xとyを図4に示します。グレーティングが回転すると、さまざまな波長の画像が出口スリットを順次通過し、光電子増倍管によって検出されます。
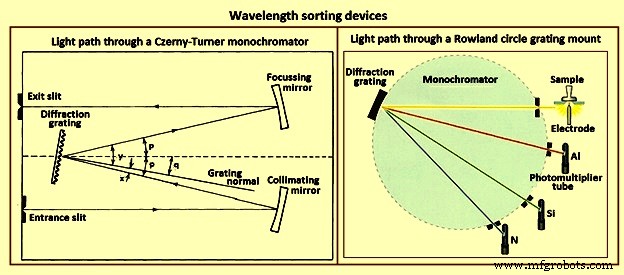
図4波長選別装置
波長ソーターの2番目の一般的なタイプは多色です。ほとんどのポリクロメーターは、ローランドサークルマウントのバリエーションです(図4)。回折格子は凹面であり、曲率半径はRです。 入口スリットが格子面に接する半径R/2の円上にある場合、スリットの回折像は円の周りに焦点を合わせます。出口スリットと光電子増倍管は、さまざまな要素からの線の波長に対応する焦点曲線上の位置に配置できます。機器の能力に応じて、40から60を超える要素までの線強度を同時に決定できます。
あるいは、フィルムのストリップまたは写真乾板を、スリットおよび光電子増倍管の代わりに焦点曲線に配置して、ポリクロメーターを分光器に変換することができます。発光スペクトル全体をプレートまたはフィルムに短時間で記録できます。写真検出により、ライン選択の柔軟性が高まり、固定スリットと光電子増倍管の組み合わせよりも多くの情報が得られます。しかし、写真媒体を処理し、関心のある線を見つけ、それらの強度を記録するために必要な時間は、写真機器の使用を面倒にする。コンピューター化されたポリクロメーターのデータ取得および処理機能の進歩により、分光機器は使用されなくなりました。
分光機器の収集光学系は、放射パワーを光源から検出器に最大の効率で伝達し、光源からの放射の空間的不均一性を解決するか、場合によってはスクランブルします。最初の要件は、光源からの放射が分光計の入口スリットとコリメート光学系を満たす場合に満たされます。適切なサイズの単純なレンズを使用して、入口スリットの光源を十分な倍率で画像化することができます。レンズのサイズは、スリットを通過する放射線がコリメート光学系をちょうど満たすように選択されます。次に、入口スリットは、システムによって見られる光源の領域を定義し、その領域内の光源の不均一性は、検出器に転送されます。写真検出では、スリット画像の空間的均一性が必要になることがよくあります。スリットの近くのレンズによって光源がコリメート光学系に画像化される場合、望ましい均一性が達成されます。次に、他のレンズを使用して、空間分解能を提供するために開口部で光源の中間画像を生成します。
排出源
放出光源は、サンプルを簡単に準備できる形から原子蒸気に分解し、次に、対象のサンプル成分から測定可能な放出信号を生成するのに十分な効率で蒸気を励起することです。 4種類の放出源(アーク、高電圧スパーク、グロー放電、および炎)のそれぞれには、分析資産と負債を伴う一連の物理的特性があります。
励起メカニズム –励起特性に最も密接に関連する放出源の特性は、温度です。温度は、ソース内のアクセス可能なエネルギーの量を示します。エネルギーはさまざまな種の間でさまざまに分割できるため、さまざまな温度がその分割を反映する可能性があります。気体の運動温度と電子の温度は、それぞれ重い粒子と電子の運動エネルギーを示しています。励起およびイオン化温度は、原子および分子種の電子エネルギー含有量を反映しています。
さらに、分子は、振動および回転温度として表される回転および振動モードでエネルギーを蓄積します。多くのソース環境では、あるモードの過剰なエネルギーが別のモードに急速に交換または転送されます。このような場合、上記の温度はすべて等しく、ソースは局所熱力学的平衡(LTE)にあります。 LTEが存在する場合、エネルギー伝達の微視的メカニズムを理解していなくても、励起条件を説明できます。与えられた種の可能な励起状態間の人口分布は、ボルツマン方程式によって与えられます。
LTEが存在しない場合、そのような場合の励起の完全な説明は、LTEを使用して予測されたものとは大きく異なる効率で特定のエネルギーレベルを励起または脱励起できる微視的な衝突プロセスを説明することです。たとえば、低圧放電では、電子集団のごく一部が放電中のガス温度よりもはるかに高い温度になる可能性があります。これらの高速電子は、LTE条件下で生成されるよりもはるかに多くの高度に励起された原子またはイオンを生成できます。非LTEソースの励起効率は、衝突する化学種の運動エネルギーまたは内部エネルギーの密接な一致に依存することが多いため、励起領域の化学組成が変化すると急激な変化を示します。
理想的な排出源 –理想的な放出源は、形状に関係なくすべての材料を効率的にサンプリングし、サンプルの組成に正比例する組成で蒸気を励起ゾーンに送ります。励起はすべての要素に対して均一に効率的です。すべての励起エネルギーがいくつかの励起状態に集中している単純なスペクトルを生成します。ソースはバックグラウンドスペクトルを生成しません。したがって、他のサンプル成分の濃度の違いに関係なく、2つのサンプルの元素濃度が等しい場合の分析結果は同じです。つまり、サンプリングと励起にはマトリックス依存性がありません。
スパークソース –高電圧スパークは、分析ギャップの自発的な破壊を引き起こすのに十分な動作電圧と、放電回路に容量的に蓄積されたエネルギーに起因する大電流を特徴とする断続的な放電です。図5は、高電圧充電回路、高電圧スイッチを備えたインダクターコンデンサータンク回路、および分析ギャップを組み込んだ波形整形回路で構成される制御波形スパークソースを示しています。
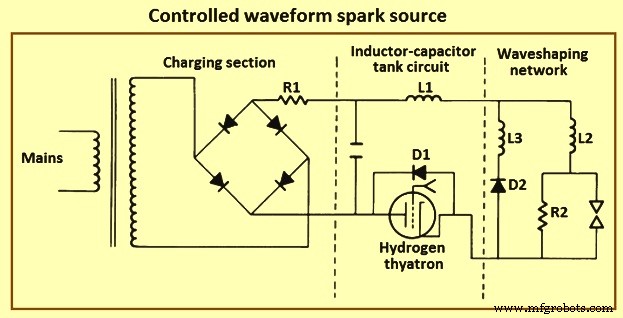
図5制御された波形スパークソース
この回路は、電流の大きさと方向、および放電時間を正確に制御して、一連の同一の火花放電を生成します。実際には、回路の充電セクションは、単に高電圧変圧器と全波整流器です。スパークは、各放電の開始時に同じコンデンサ電圧を生成するように選択された交流(AC)充電波形のゼロ交差から遅れてトリガーされます。トリガーは通常、水素サイラトロンまたは高電圧シリコン制御整流子です。与えられた放電電圧に対して、インダクタンスと静電容量の相対値は、回路のタンクと波形整形セクションの電流波形の形状と振幅を決定します。分析操作の場合、コンポーネント値は通常、ピーク振幅が50 A〜200 A、持続時間が50マイクロ秒〜150マイクロ秒の一方向放電電流を提供するように選択されます。
分析の制限 –分析用スパークギャップは、通常、分析対象の材料のタングステンピンアノードとカソードで構成されます。サンプルは電極の1つを形成するため、スパークエミッション分光法による分析は、導電性のあるサンプルまたは導電性のあるサンプルに限定されます。分析は通常、密閉されたチャンバー内またはガスの流動シースとして提供される不活性雰囲気で実行されます。安定しない限り、列車内の個々の火花はサンプル電極のさまざまな場所に当たり、平面サンプルに数ミリメートル幅の燃焼パターンを生成します。アルゴンのシースがアノードからカソードに流れると、火花がはるかに再現性よく衝突し、燃焼面積が10分の1に減少します。
スパークがサンプル電極に当たると、急速な局所加熱により電極材料がスパークギャップに放出されます。安定化されていない火花では、放出された材料の軌道はランダムです。安定した火花では、材料は電極間軸の周りの膨張シリンダーとしてギャップを通って上方に伝播します。いずれの場合も、蒸気は1回の火花の間にさまざまな励起条件にさらされます。それは最初にエネルギーのある陰極スポットを通過し、そこでいくつかの段階のイオン化を受けることができます。それが上向きに続くとき、電流伝導スパークチャネル内の蒸気は非常に励起されたままであり、電極間軸から除去された蒸気ははるかに少ないエネルギー状態を経験する。放電中の電流は増減し、ギャップを通過するサンプル蒸気の移動と一致して、励起条件を著しく変化させます。
火花の時間的および空間的不均一性は、励起温度の観点からのその特徴づけを妨げる。放出は、サンプル材料と大気ガスのイオン化のいくつかの段階から、さまざまな時間と場所で発生します。放出の主な形態は、多くの場合、最初のイオンスペクトルです。一価イオンからの線は、伝統的にスパークラインと呼ばれています。
スパーク放出の変化はマイクロ秒以内に発生します。スパークトレインからの放出も数分以内に変化します。この発光強度の長期的な変化であるスパークオフ効果は、主に、サンプル電極の表面で繰り返しスパークすることによって引き起こされるサンプル電極の変化を反映しています。電極の化学的および物理的変化は、スパークオフ効果に寄与します。したがって、スパークオフ曲線の正確な性質は、実験条件に大きく依存します。
スパークオフ動作を説明する際には、スパークソースパラメータ(静電容量、電圧、インダクタンス、繰り返し率)、サンプル組成、サンプル相構造、サンプル表面状態、スパーク雰囲気、および燃焼面積を考慮する必要があります。特に重要なのは、サンプルの組成と相構造への依存性です。これは、元素の放出結果がマトリックスに強く依存していることを示しています。これは、火花を分析放出源として使用する場合に重要です。
望ましくない特性を最小限に抑える –火花分析では、発生源の非理想性の影響を最小限に抑えるために、さまざまな手順が採用されています。スパークオフ効果は、従来、火傷に関連する最大の強度変化が発生した後にのみ放出を記録することによって処理されます。検出器への光は、通常約1分間続く予備燃焼期間中に遮断され、その間、火花が新しい電極表面を調整します。スパークが位置的に不安定で、広い領域をサンプリングする場合、ほとんどの元素について、プリバーン期間後の放出は、放出スペクトルを記録するために必要な30秒間ほぼ一定のままです。
位置的に安定した火花は、不安定な放電によって生成されるものと比較して、時間的に圧縮された火花オフ曲線を生成します。定常状態の値に増加する代わりに、発光強度は数秒以内に最大に増加し、その後比較的低い値に減少します。燃焼が続くにつれて、放出は次第に不安定になります。燃焼の最初の2分間の放出ピークには、サンプル中の元素の濃度と、元素が検出されたマトリックスのタイプに関する情報が含まれています。
火花源の非理想性に対する追加の補償は、元素濃度を示すために変更されていない放出強度ではなく強度比を使用することです。マイナーな構成要素からの線の強度は、主要なマトリックス構成要素からの強度と比例します。たとえば、鋼の分析では、合金元素からの線の強度は鉄の線の強度に比例します。この手順は、あるサンプルから別のサンプルへのサンプリングおよび励起効率の変動をいくらか補償します。これには、参照コンポーネントのサンプリングと励起が微量成分に対して同じプロセスを表すという暗黙の仮定が含まれます。これは常に当てはまるとは限りません。特に、サンプルの大部分とは濃度が大幅に異なる含有物がサンプルに含まれている場合はそうです。
上記の測定では、化学組成と物理的形態が未知のサンプルと厳密に一致する標準を使用してスパークスペクトロメータを校正しない限り、満足のいく分析結果が得られません。分析にスパークを使用するラボでは、分析する材料の種類ごとに一連の標準を用意する必要があります。火花調査の基準は簡単には作成できず、通常は購入する必要があります。
サンプリングとサンプル準備
スパークソース励起は、金属または合金の元素分析を取得するための最も迅速な方法です。サンプルは、溶鋼、薄肉、半製品または完成品から採取されます。鉄鋼業界では速度が重要であり、生産炉からの溶融物をサンプリングして分析し、合金元素の濃度を所定の範囲内に調整する必要があります。
溶鋼をサンプリングする場合は、サンプリングプローブを使用するか、サンプリングスプーンで採取した少量の溶鋼を鋳造型に流し込みます。サンプルは冷却されます。サンプルの不要な部分は、グラインダーまたはカッターを使用して除去されます。微量元素が調査されていない場合は、グラインダーまたはベルトサンダーを使用できます。平らな表面は、冷却されたサンプル上で研磨または研磨され、追加の準備なしで火花源に配置されます。ただし、汚染を最小限に抑えるために、サンプルごとに砥石またはベルトを交換する必要があります。分析が実行され、結果はすぐに炉に送られ、そこで熱の組成が適切に調整されます。サンプリングと分析には、せいぜい数分が必要です。半製品または完成品から採取したサンプルは、直径が少なくとも12mmである必要があります。小さな棒状のサンプルは、特別な装置を使用して分析できます。図6は、サンプリング、サンプル準備、および発光分光器による分析を概略的に示しています。
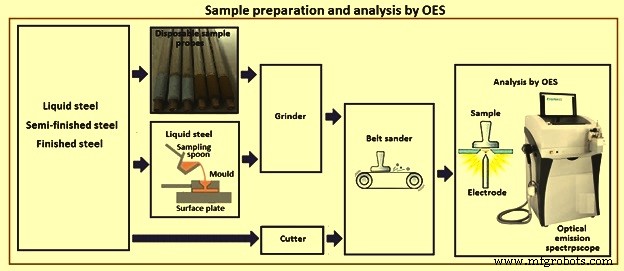
図6OESによるサンプル準備と分析
製造プロセス