


復元されたmicroRNA-133a-3pまたは枯渇したPSAT1は、GSK3β/β-カテニン経路の抑制を介して内皮細胞損傷によって誘発された頭蓋内動脈瘤を抑制します
要約
頭蓋内動脈瘤(IA)におけるマイクロRNA-133a-3p(miR-133a-3p)の機能的役割については不明です。したがって、本研究の目的は、ホスホセリンアミノトランスフェラーゼ1(PSAT1)/グリコーゲンシンターゼキナーゼ3β(GSK3β)/β-カテニンシグナル伝達を介した血管内皮損傷誘発性IAの調節に対するmiR-133a-3pの調節的役割を調査することでした。小道。正常な頭蓋内細動脈組織およびIA組織は、脳外傷およびIAの患者から収集されました。組織におけるmiR-133a-3p、PSAT1、GSK3β、およびβ-カテニンの発現は、RT-qPCRおよびウエスタンブロット分析によって決定されました。ヒトIAの内皮細胞(EC)を培養し、miR-133a-3pミミックとsi-PSAT1で処理して、内皮細胞の遊走、アポトーシス、増殖における機能を調べました。 ECにおけるmiR-133a-3p、PSAT1、GSK3β、β-カテニン、Ki-67、CyclinD1、Bax、およびBcl-2の発現は、RT-qPCRまたはウエスタンブロット分析によってテストされました。さらに、IAラットモデルは、invivoでのIA組織における病理学的変化およびmiR-133a-3p、PSAT1、GSK3β、β-カテニン、VEGF、およびMMP-9の発現を検出するために確立されました。 miR-133a-3pの発現は、IAの数とサイズに関連していた。 MiR-133a-3pの発現は死亡し、PSAT1、GSK3β、およびβ-カテニンの発現はIAで上昇しました。 miR-133a-3pを復元し、PSAT1を枯渇させると、病理学的変化が緩和されました。 IAにおけるPSAT1、GSK3β、およびβ-カテニンの発現の低下。アポトーシスを抑制し、IA ECの増殖と移動を促進し、invivoでのIA組織におけるVEGFとMMP-9の発現を減少させました。私たちの研究は、miR-133a-3pの過剰発現またはPSAT1のダウンレギュレーションが、IAのGSK3β/β-カテニン経路を阻害することにより、内皮細胞の損傷を抑制し、内皮細胞の増殖を促進することを示唆しています。 MiR-133a-3pは、IAの潜在的な候補マーカーおよび治療標的となる可能性があります。
仮説の提示
この現在の研究では、miR-133a-3p / PSAT1軸が、GSK3β/β-カテニン経路の調節を通じて内皮細胞の損傷によって誘発されるIAに影響を与える可能性があると推測できます。
仮説のテスト
この仮説を検証するために、臨床サンプル、ヒトIAの内皮細胞(EC)を収集し、IAラットモデルを確立して、IAプロセスにおけるmiR-133a-3pとPSAT1の機能を解明しました。
仮説の意味
私たちの研究は、miR-133a-3pの過剰発現またはPSAT1のダウンレギュレーションが、IAのGSK3β/β-カテニン経路を阻害することにより、内皮細胞の損傷を抑制し、内皮細胞の増殖を促進するという仮説を裏付けています。これらの発見は、IAの新しい標的療法における新しい洞察を提供します。
はじめに
頭蓋内動脈瘤(IA)は一種の脳血管障害であり、脳内の異常に膨らんだ動脈と、IA破裂によって引き起こされるくも膜下出血(SAH)を特徴とし、高い死亡率と罹患率を伴います[1]。破壊的な病気であるように、IAの病因は明らかにされていません[2]。 IAはまれな家族性の形態ですが、一般に、高血圧、喫煙、およびその他の従来の危険因子によって引き起こされた後天性血管損傷の結果であると考えられています[3]。血管内コイル塞栓術または顕微手術クリッピング術は、破裂のリスクが高い患者の未破裂動脈瘤の将来の破裂を防ぐために利用されてきました[4]。 IAの外科手術は大幅に進歩しましたが、IA患者では術後の回復が不十分です[5]。 IA治療の厳しい状況では、メカニズムをさらに調査し、新しい治療戦略を見つける必要があります。
マイクロRNA(MiRNA)は、転写後レベルでの翻訳の阻害またはmRNA分解の媒介を介して標的遺伝子の発現を調節する非コードRNAのクラスです[6]。いくつかの悪性腫瘍における腫瘍阻害剤としてのmiR-133a-3pおよびmiR-133a-3pの過剰発現は、結腸直腸癌(CRC)細胞の増殖を阻害する可能性があることが明らかになっています[7]。以前の研究では、オートファジーを介したグルタミノリシスの遮断に照らして、miR-133a-3pは胃がんの転移と増殖をさらに抑制すると主張しています[8]。また、ある研究では、miR-133a-3pが心臓の発達と心肥大の調節に関与していることが示されています[9]。ある研究では、miR-195-5pのアップレギュレーションが、ホスホセリンアミノトランスフェラーゼ1(PSAT1)依存性のGSK3β/β-カテニンシグナル伝達経路を抑制することにより、卵巣癌の血管新生とシスプラチン耐性を低下させることが示されています[10]。別の研究では、miR-365がPSAT1を調節することにより、食道扁平上皮癌(ESCC)の細胞浸潤と増殖を抑制することが報告されています[11]。 PSAT1は、セリン生合成に関与する酵素です。もともとは羊の脳から精製されており、多くの組織で高レベルになっています[12]。 PSAT1は、GSK3β/β-カテニンシグナル伝達経路の調節を介して乳がんの細胞周期進行を仲介することがわかっています[13]。 Liu etal。また、PSAT1はESCCの発生に機能を発揮し、生存率の低下を予測していることも指摘しています。したがって、それは抗癌治療の有望な標的である可能性があります[14]。前述の分析を考慮すると、この研究は、IAにおけるmiR-133a-3p / PSAT1 /GSK3β/β-カテニン軸の機能的役割のための新しいアプローチに貢献することが期待されていました。
材料と方法
倫理声明
この研究は、天津中医薬大学第一教育病院の倫理委員会によって承認されました。すべての参加者はインフォームドコンセントの文書に署名しました。すべての動物実験は、国際委員会による実験動物の管理と使用に関するガイドで集計されました。
調査対象
2016年1月から2018年3月まで、天津中医薬大学第一教育病院の脳神経外科で治療されたIAによって引き起こされたSAHの症例が選択されました。顕微手術によって得られた75例のIAの病理学的サンプルが収集され、平均年齢44.98±6.79歳の31〜55歳の男性29人と女性46人を含むIAグループとして分類されました。天津中医薬大学第一教育病院で同時に脳神経外科で治療された脳外傷の患者が対照群として選ばれた。また、正常な頭蓋内細動脈組織の75例は、外傷性手術または内部減圧によって蓄積され、34〜56歳の男性43人と女性32人を含み、平均年齢は48.14±8.68歳でした。高血圧、糖尿病、または腫瘍の病歴がある患者は除外されました。 IA群と対照群(両方 P )の間で性別と年齢に顕著な違いはありませんでした。> 0.05)。
サンプルの処理と保存
外科的切除後、2つのグループのサンプルの一部をホルムアルデヒドで固定し、低濃度から高濃度までの勾配アルコールで脱水し、パラフィンで包埋しました。次に、サンプルをヘマトキシリン-エオジン(HE)染色および免疫組織化学的染色のためにスライスした。一部のサンプルは液体窒素タンクにすばやく入れられ、ウエスタンブロット分析および逆転写定量的ポリメラーゼ連鎖反応(RT-qPCR)の検出のために-80°Cの極低温冷蔵庫に移されました。一部のサンプルは電子顕微鏡観察用にグルタルアルデヒドで固定され、一部のサンプルはECの分離に利用されました。
電子顕微鏡観察
サンプルを3%グルタルアルデヒドで固定した後、1%四酸化オスミウムで再固定しました。サンプルをアセトンで脱水し、Epon812を埋め込んで、厚さ3μmの半薄切片にスライスしました。最後に、サンプルを酢酸ウラニルとクエン酸鉛で二重染色し、H-600IV透過型電子顕微鏡(日立、東京、日本)で観察しました。
HE染色
調製したパラフィン切片を60°Cで30分間焼きました。上記の手順が完了した後、組織スライスをキシレンで固定し、勾配無水アルコールで脱水し、リン酸緩衝生理食塩水(PBS)で洗浄しました。組織スライスをヘマトキシリンで染色し、アンモニアで数秒間処理し、エオシンで2分間染色し、脱水し、透明にしました。次に、組織スライスに中性ガムを滴下し、カバーガラスで密封した。最後に、顕微鏡(ニコン、東京、日本)が観察と記録に採用されました。
免疫組織化学的染色
免疫組織化学キットは、Zymed Laboratories(サンフランシスコ、カリフォルニア州、米国)によって製造されました。パラフィンスライスを脱ロウおよび水和し、パラフィンスライスをキシレン溶液に5分×3回浸漬しました。スライスを100%無水アルコールに3分間×2回入れ、次に95〜75%アルコールに3分間浸しました。脱ろう後、スライスを3%過酸化水素で15分間インキュベートして、内因性ペルオキシダーゼの活性を排除しました。スライスにブロッキング溶液を滴下し、通常のヤギ血清作業溶液と15分間インキュベートした後、マトリックスメタロプロテアーゼ(MMP)-9(5μg/ mL)および血管内皮増殖因子(VEGF)(1:250)に対する一次抗体でプローブしました。 、アブカム、ケンブリッジ、マサチューセッツ、米国)(ネガティブコントロール(NC)用のPBS)および1〜2時間インキュベート。スライスをビオチン化二次抗体ワーキング溶液で30〜60分間再プローブしました。スライスに、西洋ワサビペルオキシダーゼで標識されたストレプトアビジン/ペルオキシダーゼ作動油を加え、新しく調製したジアミノベンジジン(DAB)溶液を滴下し、対比染色し、ブロックしました。この画像は、Nikon SPOTFlexTMイメージングシステムによって認識されました。 MMP-9およびVEGFタンパク質発現の領域は、免疫組織化学的定量分析ソフトウェアによって測定されました。各サンプルの陽性細胞の蓄積領域で5つの高倍率視野がランダムに検出され、各画面の平均吸光度が統計分析の平均値として使用されました。
ECの分離と文化
ECは、正常な頭蓋内細動脈組織およびIA組織から分離され、培養されました。組織を3mm 2 にスライスしました 断片化し、0.1%コラゲナーゼB / 0.1%ジスパーゼ(Roche、バーゼル、スイス)で25分間インキュベートしました。組織を剥がし、2 mLピペットで2分間粉砕し、100μmストレーナー(BD Biosciences、NJ、USA)でろ過してECを分離しました。細胞懸濁液を遠心分離した後、成長因子と20%ウシ胎児血清(PromoCell、ハイデルベルク、ドイツ)を含む培地MV2に再懸濁しました。次に、細胞をフィブロネクチン(Sigma Aldrich Inc.、米国ミズーリ州セントルイス)でコーティングされた皿に10 4 の密度で播種しました。 セル/ cm 2 (1μg/ cm 2 )、5%CO 2 で1日間成長させます 。播種の翌日、細胞をPBSでリンスして付着していない細胞を除去し、新しい培地に入れました。約80〜100%のコンフルエンスに達したとき、培養物はUlex europaeus Agglutinin I(UEA)コーティング(Vector Laboratories、Ltd.、Peterborough、UK)ビーズ(Dynabeads M-450 Tosylactivate、Oxoid、Hampshire、UK)による免疫分離にさらされました。純粋なECを取得します。レクチンでコーティングされたビーズに結合したECを磁性粒子濃縮器で集め、結合していない細胞を基本培地で2回洗浄して除去しました。 UEA陽性細胞を培地に再懸濁し、フィブロネクチンでコーティングした皿に播種して、接着と増殖を改善しました。文化は4〜6日以内にコンフルエントになりました。
ECの識別
ECは、細胞表面CD31抗体とFVII因子関連抗原を用いた免疫細胞化学的染色によって同定されました。細胞をPBSで2回洗浄し、4%パラホルムアルデヒドで固定し、3%H 2 でインキュベートしました。 O 2 内因性ペルオキシダーゼ活性を除去するために10〜15分間、次に0.1%TritonX-100と10分間インキュベートして穴の開いた細胞にします。細胞に特定の一次抗体:第VII因子(1:200)、CD31(1:400、ロシュ、バーゼル、スイス)を滴下し、4°Cで一晩インキュベートしました。次に、西洋ワサビペルオキシダーゼ二次抗体で標識した免疫グロブリンG(1:50)を細胞に滴下しました。細胞を37°Cで45分間インキュベートし、DABが4分間光を避けて発育させました。その後、蒸留水で発色を停止し、顕微鏡で写真を観察した。細胞を蛍光倒立位相差顕微鏡下で観察し、陽性細胞と細胞の総数を10視野からランダムに数えた。陽性染色細胞率=(陽性細胞数/細胞総数)×100%。対応するNCグループが確立され、一次抗体がPBSに置き換えられ、他のステップは上記のように実行されました。
細胞のグループ化とトランスフェクション
IAのECに対するmiR-133a-3pとPSAT1の効果を研究するために、ECをコントロールグループ(トランスフェクションなしの正常な血管EC)、IAグループ(トランスフェクションなしのIA血管EC)、模倣NCグループ(miRでトランスフェクト)にグループ化しました。 -133a-3pミミックNC)、miR-133a-3pミミックグループ(miR-133a-3pミミックでトランスフェクト)、低分子干渉RNA(si)-NCグループ(si-PSAT1 NCでトランスフェクト)、si-PSAT1グループ(トランスフェクトsi-PSAT1)、およびmiR-133a-3pミミック+過剰発現(oe)-PSAT1グループ(miR-133a-3pミミックおよびoe-PSAT1でトランスフェクト)。その中で、mimic NC、miR-133a-3p mimic、si-PSAT1、si-NC、およびoe-PSAT1は、GenePharma Co.、Ltd。(上海、中国)によって考案および作成されました。トランスフェクションは、Lipofectamine TM の指示に厳密に従って実施されました。 2000トランスフェクション試薬(Thermo Fisher Scientific、マサチューセッツ、米国)。
フローサイトメトリー
培養皿中の培地を廃棄し、細胞をPBSで2回すすいだ。細胞を0.25%トリプシンで剥離し、800 rpmで5分間遠心分離し、1×結合バッファーで懸濁し、細胞密度を1×10 7 に調整しました。 細胞/ mL。細胞懸濁液(100μL)を5μLのヨウ化プロピジウム(PI、20μg/ mL)およびアネキシンV-FITCと20分間インキュベートした後、400μLの1×結合バッファーと混合しました。フローサイトメーター(BD FACSArial Iセルソーター)を使用して、1時間以内に細胞アポトーシスを検出しました。結果は、散布図の左下の象限(Q4)が健康な生細胞(FITC - )を示したことです。 / PI − )、初期アポトーシス細胞(FITC + )としての右下象限(Q3) / PI − )、および右上の象限(Q2)は、後期アポトーシス細胞およびアポトーシス細胞(FITC + )でした。 / PI + );アポトーシス率=初期アポトーシス率(Q3)+後期アポトーシス率(Q2)。
3-(4,5-ジメチルチアゾール-2-イル)-2,5-ジフェニルテトラゾリウムブロミド(MTT)アッセイ
細胞をトリプシンで剥離して細胞懸濁液を調製した。倒立顕微鏡下で細胞を数えた。細胞濃度を5×10 4 に調整しました 細胞/ mL。細胞を96ウェル培養プレートに播種しました。 48時間後、細胞を20μLのMTT溶液で4時間インキュベートしました。各ウェルのMTTを150μLのジメチルスルホキシドで溶解しました。 ECの光学密度(OD)値は、570nmの波長で測定されました。 ECの増殖率は、OD値に照らして計算されました。
スクラッチテスト
各グループの細胞は、2×10 5 の24ウェルプレートに播種されました。 細胞/ウェル。各グループに3つの平行なウェルを設定しました。約90%のコンフルエンスに達したとき、細胞増殖面を滅菌済みの使い捨て1mLマイクロピペットチップで引っ掻きました。各ウェルに1回引っかき傷を付け、各ウェルの引っかき傷の長さと深さは一定でした。スクラッチ後、浮遊細胞を除去し、培地を新しいものと交換し、24時間の培養後に顕微鏡下でスクラッチ間隔を観察した。引っかき傷の治癒領域は、National Instrument Vision Assistant8.6ソフトウェアによって列挙されました。細胞移動=創傷治癒領域/最初の引っかき傷領域×100%。
実験動物とIAラットモデルの確立
7週齢で体重が180〜200 gの84匹のSprague-Dawley(SD)ラット(軍事医学研究院、実験動物センター、北京、中国)を選択しました。ラットは動物実験センターに収容された。給餌条件は、自然光で22〜25°C、湿度50〜60%に制御されました。すべてのラットは、ケージあたり4匹のラットを含む標準的なラットケージで飼育された。ラットは都市の衛生的な飲料水と一般的なラットの飼料を与えられました。クッションは3日ごとに交換し、ケージを洗浄・滅菌しました。 IAラットは参考文献[15]に従ってモデル化されました。動脈瘤破裂は、ラットで次の症状が発生したときに確認されました[16] 。1、24時間にわたる体重減少(約10%の体重減少)によって行われた飲食活動の低下。 2、持ち上げる際の胴体と前肢の屈曲。 3、通常の姿勢で片側を歩く。 4、安静時に片側に寄りかかって、自発的な活動はありません。これらの症状のあるラットは、手術の3か月後に安楽死させた。手術中にIA組織を採取し、PBSで灌流し、グルタミン酸を含む青色色素を大脳動脈に灌流しました。
IAラットの治療と介入
上記の84匹のラットをランダムに7つのグループに分け、各グループに12匹のラットを入れました。治療法は以下の通りであった:正常群(モデリングは行われなかった)。 IAグループ(PBSとLipofectamine2000の100μL混合物を注射した定位固定);模倣NCグループ(miR-133a-3p模倣NCとリポフェクタミン2000の100μL混合物による定位注射); miR-133a-3pミミックグループ(miR-133a-3pミミックとLipofectamine 2000の100μL混合物による定位注射); si-NCグループ(si-PSAT1NCとLipofectamine2000の100μL混合物による定位注射); si-PSAT1グループ(si-PSAT1とLipofectamine 2000の100μL混合物による定位注射);およびmiR-133a-3pミミック+ oe-PSAT1グループ(miR-133a-3pミミックとoe-PSAT1およびLipofectamine 2000の100μL混合物による定位注射)。上記の注射はすべて1日1回行われ、これらのラットは特定病原体除去(SPF)動物実験室で12週間飼育されました。 12週間後、各群のラットに麻酔をかけ、上記のように胸腔を開いた。大動脈に挿管している左心室から、大静脈を切断することによって血液が放出された。同時に、ヘパリンナトリウム(37°C)を含む生理食塩水30 mLを管から灌流し、10%ポリホルムアルデヒド/0.1Mリン酸緩衝液(pH 7.4)を管からゆっくりと脳に注入しました。灌流が固定された後、脳が開かれた。頭蓋底の動脈輪を手術顕微鏡下で分離・除去し、動脈瘤の変化を顕微鏡下で観察し、病理学的特徴を調査した。 mimic NC、miR-133a-3p mimic、si-NC、si-PSAT1、およびoe-PSAT1は、Shanghai Sangon Biotechnology Co.、Ltd。(上海、中国)によって配合されました。
血行動態の検出
ラットの左総頸動脈の末端の血液の流量を、手術の3日前と介入治療の12週間後にテストしました。方法は次のとおりです。ラットを麻酔器の動物フレームに入れて吸入し、フローパラメータを調整しました。ラットが安定して呼吸し、ラットの尾に触れても明らかな反応がなかった後、ラットを輪ゴムで実験手術台に固定した。ラットの首の毛を電気かみそりで剃った。カラードップラー超音波検出器をオンにし、左総頸動脈の端の血流速度を測定し、プローブを適切なカップリング剤で塗った後にデータを記録した。測定後、麻酔後にラットが目覚めるまで気道を塞がないように、ラットを慎重にケージに戻しました。
RT-qPCR
全RNAは、RNA単純全RNA抽出キット(TIANGEN Biotechnology Co.、Ltd。、北京、中国)に基づいて抽出されました。高品質のRNAは、紫外線分析とホルムアルデヒド変性電気泳動によって確認され、RNAはPrimeScriptRT試薬キットによって相補DNAに逆転写されました。 PCR反応はSYBRPermix Ex Taq II によって実施されました。 (タカラ、大連、遼寧、中国)。 PCRプライマーは、Beijing ComWin Biotech Co.、Ltd。(北京、中国)によって考案および配合されました(表1)。 U6は、miR-133a-3p、PSAT1、GSK3β、β-カテニン、Bax、Bcl-2、Ki-67、およびCyclinD1の内部パラメーターとして選択され、内部パラメーターとしてグリセルアルデヒドリン酸デヒドロゲナーゼ(GAPDH)が使用されました。データは2 -ΔΔCt によって測定されました 。
<図>ウエスタンブロット分析
総タンパク質は細胞と組織から抽出され、タンパク質サンプルはビシンコニン酸タンパク質アッセイキット(Beyotime Institute of Biotechnology、上海、中国)によって定量化されました。サンプルを1/4容量の5×サンプルバッファーと混合し、5分間煮沸しました。電気泳動には10%分離ゲルと5%濃縮ゲルを選択しました。膜を5%脱脂粉乳で60分間孵化させた。メンブレンに一次抗体PSAT1(1:500)、GSK3β(1:500)、β-カテニン(1:5000)、Bax(1:1000)、Bcl-2(1:1000)、CyclinD1(1:1)を付加しました。 200)、Ki-67(1:5000)、MMP-9(1μg/ mL)、VEGF(1:1000)(すべてAbcam、ケンブリッジ、マサチューセッツ、米国から)。次に、メンブレンを二次抗体(1:2000)で60分間ハッチングしました。メンブレンをエレクトロケミルミネッセンス反応溶液(Beyotime Institute of Biotechnology、上海、中国)に1分間浸し、液体を除去した後、フードラップで覆った。メンブレンをX線で露光し、現像して固定した後に結果を観察しました。 GAPDH(1:10000、Abcam)をローディングコントロールとして使用し、タンパク質画像をImageJ2xソフトウェアで分析しました。
デュアルルシフェラーゼレポーター遺伝子アッセイ
miR-133a-3pとPSAT1の間のターゲット関係、およびmiR-133a-3pとPSAT1 3 '非翻訳領域(3'UTR)の間の結合部位は、バイオインフォマティクスWebサイト(https://cm.jefferson.edu/rna22)によって予測されました。 / Precomputed /)。 miR-133a-3p結合部位を含むPSAT13'UTRプロモーター領域の配列を増幅し、pGL3-basic luciferaseプラスミド(Takara Bio Inc.、Otsu、Shiga、Japan)にクローニングして、野生型(WT)プラスミド(WT)を構築しました。 PSAT1 3'UTRのPSAT1-WT)、変異型(MUT)PSAT1-MUT組換えプラスミドは、PSAT1-WT上のmiR-133a-3p結合部位を点突然変異キット(タカラバイオ株式会社、大津、志賀)で突然変異させることによって処方されました。 、 日本)。対数増殖期の血管ECを96ウェルプレートに播種しました。コンフルエンスが約70%に達したときに、PSAT1-WTおよびPSAT1-MUTプラスミドをLipofectamine 2000によって模倣NCおよびmiR-133a-3p模倣プラスミドと混合し、血管ECにコトランスフェクトしました。トランスフェクションの48時間後に細胞を回収して溶解し、ルシフェラーゼ検出キット(Promega Corporation、米国ウィスコンシン州マディソン)でルシフェラーゼ活性を確認しました。
統計分析
すべてのデータは、SPSS 21.0ソフトウェア(IBM Corp.、米国ニューヨーク州アーモンク)によって説明されました。列挙データはレートまたはパーセンテージで示され、分析はカイ2乗検定またはフィッシャーの直接確率検定によって決定されました。正規分布の測定データは、平均±標準偏差で表されました。 2つのグループ間の比較は t によって行われました。 複数のグループ間の比較は、一元配置分散分析(ANOVA)とそれに続くテューキーの事後検定によって分析されました。 P 0.05未満の値は有意であると見なされました。
結果
IA患者の一般データ
表2に示すように、IA群と対照群の一般的なデータを比較しました。具体的な情報を表2に示します。
<図>miR-133a-3pの表現はIAの数とサイズに関連しています
miR-133a-3p発現とIAの臨床病理学的特徴との関係を分析することにより、IAにおけるmiR-133a-3pの平均相対発現に照らして、75例のIAが2つに分布したことが表3に詳述された。グループ:miR-133a-3p高発現グループ( n =47)およびmiR-133a-3p低発現グループ( n =28)。 miR-133a-3pとさまざまな臨床病理学的パラメーターとの関係は、カイ2乗検定またはフィッシャーの直接確率検定によって統計的に分析されました。結果は、miR-133a-3pの発現が動脈瘤の年齢、性別、形状、および位置に関連していないことを示しました(すべて P > 0.05)、ただし動脈瘤の数とサイズに関連している( P の両方) <0.05)。
<図>MiR-133a-3pの発現が低下し、PSAT1、GSK3β、およびβ-カテニンの発現がIA組織で上昇する
IAにおけるmiR-133a-3pの発現はRT-qPCRによって決定され、その結果、正常な頭蓋内細動脈組織(対照群)と比較して、miR-133a-3pの発現がIA組織( IAグループ)( P <0.05)(図1a)。 RT-qPCRおよびウエスタンブロット分析により、PSAT1、GSK3β、およびβ-カテニンの発現が、正常な頭蓋内細動脈組織(すべて P )と比較してIA組織で上昇したことが明らかになりました。 <0.05)(図1a–c)。
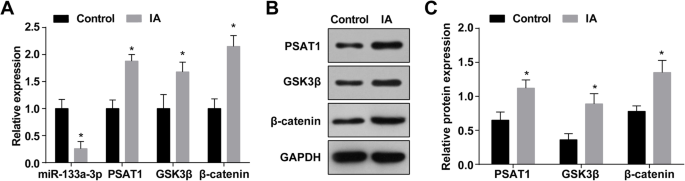
IA組織では、MiR-133a-3pの発現が減少し、PSAT1、GSK3β、およびβ-カテニンの発現が増加します。 a IA群および正常群におけるmiR-133a-3p、PSAT1、GSK3β、およびβ-カテニンの発現。 b PSAT1、GSK3β、およびβ-カテニン発現のタンパク質バンド。 c ウエスタンブロットアッセイによるIA群および正常群におけるPSAT1、GSK3β、およびβ-カテニンのタンパク質発現。 n =75、* P <0.05対対照群。測定データは平均±標準偏差として表され、2つのグループ間の比較は独立したサンプル t によって行われました。 テスト
IA組織における動脈瘤とMMP-9およびVEGF発現の病理学的変化
正常な頭蓋内細動脈組織およびIA組織を直接観察することにより、対照群では、動脈組織の血管は真っ赤であり、内腔に明らかなアテローム硬化性プラークおよび外側血栓は見られなかったことが示された。 IA群の動脈瘤組織の腫瘍は、ほとんどが褐色または暗赤色であり、外観は陰茎状または紡錘状であり、組織はほとんど硬いものであった。腫瘍を切り開くと、一部の腫瘍サンプルの腫瘍壁に白または暗赤色のアテローム性動脈硬化症のプラークが現れ、平ら、円形、または楕円形でした。一部の腫瘍サンプルでは、腫瘍腔に壁の血栓があり、血栓のテクスチャーは柔らかかった。腫瘍の壁の厚さは腫瘍の首から徐々に薄くなり、腫瘍の上部に薄い繊維膜しかないものもあれば、破裂したものもありました。破裂した動脈瘤のクレバスは、腫瘍の上部またはその近くにありました。
HE染色は、光学顕微鏡下で、正常な頭蓋内細動脈組織の壁の厚さが均一であることを示した。内層、中層、外層の解剖学的構造は明確で無傷でした。各層の細胞の形態は正常でした。隣接する細胞の筋鞘はしばしば密接に形成された。そして壁の炎症細胞はまれでした。 IA群では、動脈瘤壁の頭蓋内動脈分岐部上部の遠位外側血管腔に形成された局所突起が鈍く小さくなり、局所ECが失われた。少数のサンプルは、平滑筋細胞層から内膜層への移動および筋形成内膜細胞増殖を示した。 ECは減少するか、さらには消失しました。内皮細胞層は、過形成性筋内膜細胞と直線的に配置されたEC、または内腔に付着したアポトーシスECと血液細胞によって構成されていました。その液胞は退化し、連続性の中断を示した。それらのいくつかは基底膜と一緒に剥がれ、内膜コラーゲン線維が増加しました。アテローム性動脈硬化症は変化し、細動脈壁は明らかに薄く、多数の結合ラダー組織で満たされていました。炎症性細胞の浸潤と部分的な拡散が、主に中膜と外膜のすべての層で観察されました。一部の細胞では脂質とコレステロールの結晶沈着が観察された。一部の腫瘍壁は完全にまたは局所的に薄くなり、外側に拡張しました(図2a、b)。
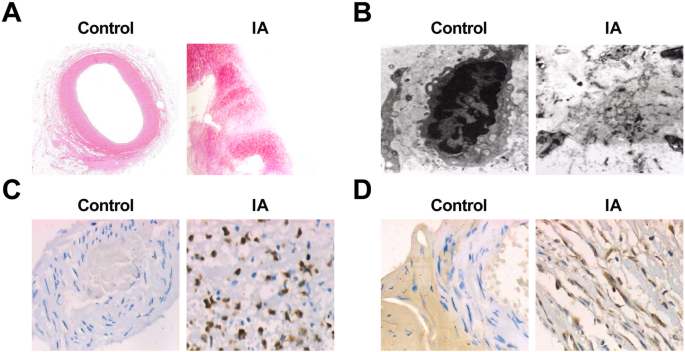
動脈瘤の病理学的変化およびIAにおけるMMP-9およびVEGFの発現。 a HE染色下の対照群の正常な頭蓋内細動脈組織切片(×10)。 b HEで染色したIA組織切片(×10)。 c 電子顕微鏡下での対照群の正常な頭蓋内細動脈組織の超微細構造(×10,000)。 d 電子顕微鏡下でのIA組織の超微細構造(×10,000)。 e 免疫組織化学的染色による対照群およびIA群におけるMMP-9の発現(×200)。 f 免疫組織化学的染色による対照群およびIA群におけるVEGFの発現(×200)
正常な頭蓋内細動脈組織とIA組織の切片を電子顕微鏡で観察したところ、正常な頭蓋内細動脈組織では脳血管壁のマトリックス線維がはっきりと見え、内皮損傷や細胞核濃縮は見られなかった。 、または退化。 IA組織では、明らかな内皮細胞損傷、細胞核濃縮、または液胞変性が観察され、中平滑筋細胞の数が減少し、核濃縮のほとんどが現れ、クロマチン凝集およびアポトーシス小体が見られた。一部の細胞はミトコンドリアの腫れと正常な内部構造の消失を示しました。細胞骨格を形成する細胞外マトリックスはぼやけており、無定形のフロックを示した。細胞の欠けている部分に多くの断片がありました(図2c、d)。
免疫組織化学的染色を利用して、MMP-9およびVEGFの発現を試験し、その結果、対照群の75例でMMP-9およびVEGFの発現がないことが明らかになった。 IAサンプル75例にMMP-9の陽性発現が60例存在した。 MMP-9陽性発現はIA壁の内膜と外膜に現れたが、発現は均一ではなかった。陽性発現は主に茶色がかった黄色の細胞質によって特徴づけられた。 VEGFの陽性発現は、IAサンプルの75例で66例であった。 IAの壁では、中膜および外膜で高い陽性発現があり、内膜で低い陽性発現があった。陽性発現はまた、主に茶色がかった黄色の細胞質によって特徴づけられた(図2e、f)。 2つのグループにおけるMMP-9とVEGFの発現を表4に示します。
<図>血管ECの識別
ECにおける第VIII因子およびCD31の発現は、免疫組織化学的染色によって分析された。その結果、血管ECは第VIII因子およびCD31関連抗原抗体に陽性反応を示し、陽性率は95%でした。さらに、細胞質には多数の褐色粒子があり、褐色染色の細胞の5回目の継代は、細胞の1回目の継代よりも劇的に高かった(図3a、b)。
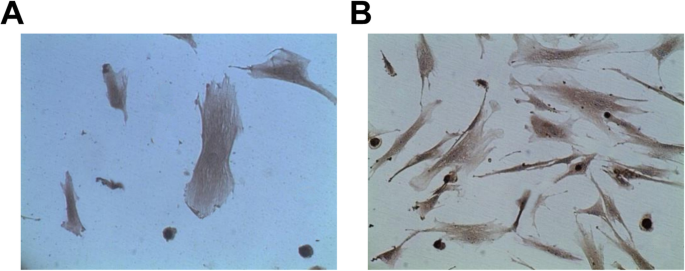
血管ECは、FVIIIおよびCD31関連の抗原抗体に積極的に反応します。 a CD31によるECの識別。 b FVIIによるECの同定
miR-133a-3pのアップレギュレーションとPSAT1のダウンレギュレーションは、アポトーシスを抑制し、IAにおけるECの増殖と移動を促進します
フローサイトメトリー、RT-qPCR、およびウエスタンブロット分析を採用して、miR-133a-3pミミックまたはsi-PSAT1で処理した後のIAのECにおけるアポトーシスとBaxおよびBcl-2の発現を観察しました。対照群と比較して、IA群では細胞のアポトーシス率とBax発現が上昇し、Bcl-2発現が低下したことが示された(すべて P <0.05)。 IAグループ、模倣NCグループ、およびsi-NCグループの細胞アポトーシスとBaxおよびBcl-2の発現には、有意な変化はありませんでした(すべて P > 0.05)。 si-NC群および模倣NC群と比較すると、si-PSAT1群およびmiR-133a-3p模倣群の細胞のアポトーシス率が抑制され、Bax発現が低下し、Bcl-2発現が上昇した(すべての P <0.05)。 miR-133a-3p模倣グループと比較して、アポトーシス率とBax発現は増強され、Bcl-2発現はmiR-133a-3p模倣+ oe-PSAT1グループ(すべて P )で減少しました。 <0.05)(図4a–e)。
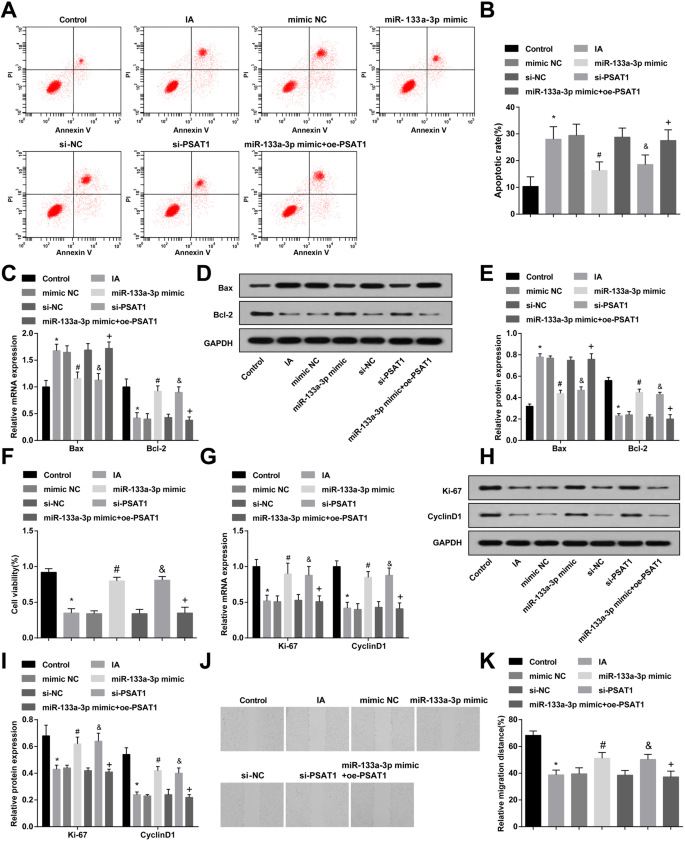
高発現のmiR-133a-3pと低発現のPSAT1はアポトーシスを阻害し、IAECの増殖と移動を促進します。 a フローサイトメトリーによるECのアポトーシスの検出。 b 各グループのECのアポトーシス率の検出。 c RT-qPCRによって検出されたECでのBaxおよびBcl-2の発現。 d BaxおよびBcl-2発現のタンパク質バンド。 e ウエスタンブロット分析によって検出されたECにおけるBaxおよびBcl-2タンパク質の発現。 f MTTアッセイは、各グループのECの増殖活性を検出するために使用されました。 g RT-qPCRを使用して、ECの各グループでKi-67とCyclinD1の発現を検出しました。 h Ki-67のタンパク質バンドとCyclinD1の発現。 i ウエスタンブロット分析によって検出されたECにおけるKi-67およびCyclinD1タンパク質の発現。 j スクラッチテストによる各グループのECの移行の検出。 k 各グループにおける内皮細胞遊走の統計結果。 N =3、* P <0.05対対照群。 # P <0.05vs。模倣NCグループ。 & P <0.05 vs.si-NCグループ。 + P <0.05 vs.miR-133a-3pミミックグループ。測定データは平均±標準偏差として表されました。データは、一元配置分散分析とそれに続くテューキーの事後検定によって評価されました
MTTアッセイ、RT-qPCR、およびウエスタンブロット分析を利用して、miR-133a-3pミミックまたはsi-PSAT1で処理した後のIAのECにおけるKi-67およびCyclinD1の増殖と発現を観察しました。対照群とは対照的に、増殖活性とKi-67およびCyclinD1の発現はIA群で減少したことが示されました(すべて P <0.05)。 IAグループ、模倣NCグループ、およびsi-NCグループ(すべて P )の増殖活性とKi-67およびCyclinD1の発現に有意差はありませんでした。> 0.05)。 si-NCグループと模倣NCグループに関連して、増殖活性とKi-67およびCyclinD1発現は、si-PSAT1グループとmiR-133a-3p模倣グループ(すべて P > <0.05)。 miR-133a-3p模倣グループと比較して、miR-133a-3p模倣+ oe-PSAT1グループ(すべて P )では、増殖活性とKi-67およびCyclinD1の発現が減少しました。 <0.05)(図4f、i)。
miR-133a-3pミミックまたはsi-PSAT1で24時間処理した後の各グループのECの移動は、スクラッチテストによって観察されました。 IA群の細胞の遊走は対照群の細胞の遊走と比較して阻害されたことが明らかになった( P <0.05)。 IAグループ、si-NCグループ、および模倣NCグループ(すべて P )の細胞移動に顕著な変化はありませんでした。> 0.05)。 si-NCグループおよび模倣NCグループと比較して、si-PSAT1グループおよびmiR-133a-3p模倣グループの細胞移動は上昇しました(両方 P <0.05)。 miR-133a-3p模倣グループと比較して、miR-133a-3p模倣+ oe-PSAT1グループ( P )では細胞遊走が低下しました。 <0.05)(図4j、k)。
復元されたmiR-133a-3pと枯渇したPSAT1は、IAのECにおけるPSAT1、GSK3β、およびβ-カテニンの発現を低下させます
RT-qPCRを使用して、IAのECにおけるmiR-133a-3pの発現を検出しました。対照群と比較して、IA群のmiR-133a-3p発現が減少したことが得られた( P <0.05)。 IAグループ、模倣NCグループ、およびsi-NCグループでのmiR-133a-3pの発現は、著しく変化しませんでした( P > 0.05)。 miR-133a-3pミミックグループのMiR-133a-3p発現は、ミミックNCグループの発現と比較して増強されました( P <0.05)。 si-NCグループとは対照的に、si-PSAT1グループ( P )ではmiR-133a-3pの発現に明確な変化はありませんでした。> 0.05)。 miR-133a-3p模倣グループと比較して、miR-133a-3p発現は、miR-133a-3p模倣+ oe-PSAT1グループ( P )で有意差を示さなかった。> 0.05)(図5a)。
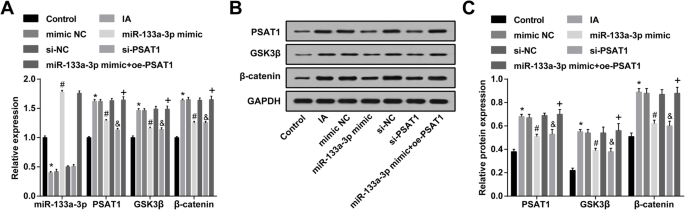
miR-133a-3pのアップレギュレーションとPSAT1のダウンレギュレーションは、IAのECにおけるPSAT1、GSK3β、およびβ-カテニンの発現を減少させます。 a RT-qPCRによって検出されたECにおけるmiR-133a-3p、PSAT1、GSK3β、およびβ-カテニンの発現。 b PSAT1、GSK3β、およびβ-カテニンのタンパク質バンド。 c ウエスタンブロット分析によって検出された各グループのECにおけるPSAT1、GSK3β、およびβ-カテニンタンパク質の発現。 N =3、* P <0.05対対照群。 # P <0.05vs。模倣NCグループ。 & P <0.05 vs.si-NCグループ。 + P <0.05 vs.miR-133a-3pミミックグループ。測定データは平均±標準偏差として表され、データは一元配置分散分析とそれに続くテューキーの事後検定によって評価されました
IAのECにおけるPSAT1、GSK3β、およびβ-カテニンの発現は、ウエスタンブロット分析およびRT-qPCRによってテストされました。対照群と比較して、IA群のPSAT1、GSK3β、およびβ-カテニンの発現が上昇したことが示された(すべての P <0.05)。 IAグループ、模倣NCグループ、およびsi-NCグループにおけるPSAT1、GSK3β、およびβ-カテニンの発現は劇的に変化しませんでした(すべての P > 0.05)。 miR-133a-3pミミックグループおよびsi-PSAT1グループのPSAT1、GSK3β、およびβ-カテニンの発現は、ミミックNCグループおよびsi-NCグループの発現と比較して低下しました(すべて P <0.05)。 miR-133a-3pミミックグループに関連して、PSAT1、GSK3β、およびβ-カテニンの発現は、miR-133a-3pミミック+ oe-PSAT1グループ(すべて P )で上昇しました。 <0.05)(図5a–c)。
miR-133a-3pのアップレギュレーションとPSAT1のダウンレギュレーションは、IA組織の病理学的変化を軽減します
モデリング後のラットの血行力学的変化をテストすることにより、手術の3日前と介入治療の12週間後に各グループのラットの血流速度を監視しました。手術の3日前に各群で血流速度に明らかな差はないことが行われた( P > 0.05)。介入の12週間後、IAグループのラットの血流速度は正常グループのそれと比較して低下しました( P <0.05)。 IA群、模倣NC群、si-NC群、miR-133a-3p模倣+ oe-PSAT1群(すべて P )では、血流速度の低下の程度に明確な違いはありませんでした。> 0.05)。 si-NC群および模倣NC群と比較すると、miR-133a-3p模倣群およびsi-PSAT1群( P )で血流速度が上昇した。 <0.05)。 miR-133a-3p模倣群とは対照的に、miR-133a-3p模倣+ oe-PSAT1群では血流速度が低下しました( P <0.05)(図6a)。
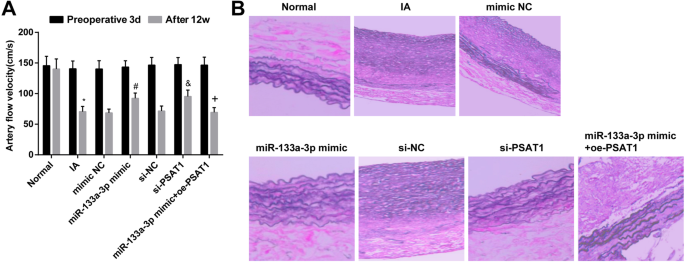
ダウンレギュレーションされたmiR-133a-3pとダウンレギュレーションされたPSAT1は、IA組織の病理学的変化を軽減します。 a ラットでのモデリングが成功した後の各時点での血行力学的変化。 b トランスフェクション後のIA組織の変化。 n =12、* P <0.05対通常のグループ。 # P <0.05vs。模倣NCグループ。 & P <0.05 vs.si-NCグループ。 + P <0.05 vs.miR-133a-3pミミックグループ。測定データは平均±標準偏差として表され、データは一元配置分散分析とそれに続くテューキーの事後検定によって評価されました。
IA組織の変化はHE染色によって確認されました。その結果、正常群では頭蓋内維管束組織中層の弾性線維がきれいで、正常な弾性タンパク質の波状構造が現れ、破損や劣化は見られなかった。正常群と比較して、頭蓋内血管組織の内腔が拡大し、正常な弾性タンパク質の波状構造が消失し、局所弾性タンパク質血管の中層の弾性線維が破壊され、一部の弾性線維が完全に破壊された。 IAグループで劣化しました。 si-NCグループ、mimic NCグループ、IAグループ、およびmiR-133a-3p mimic + oe-PSAT1グループのIA組織の形態に明確な変化はありませんでした。 si-NC群および模倣NC群とは対照的に、miR-133a-3p模倣群およびsi-PSAT1群のラットの頭蓋内血管組織における弾性タンパク質の波形構造が存在し、局所弾性タンパク質血管が存在した。構造はわずかに無秩序でしたが、破壊と溶解はありませんでした(図6b)。
高発現miR-133a-3pおよび低発現PSAT1は、生体内のIA組織におけるPSAT1、GSK3β、β-カテニン、VEGF、およびMMP-9の発現を低下させます
インビボでのIA組織におけるmiR-133a-3pの発現は、RT-qPCRによって試験され、正常群と比較して、miR-133a-3p発現は、IA群において低下したことが示唆された(<i> P <0.05)。 IAグループ、模倣NCグループ、およびsi-NCグループでのmiR-133a-3pの発現は、明らかに変化しませんでした(すべての P > 0.05)。 miR-133a-3pミミックグループのMiR-133a-3p発現は、ミミックNCグループの発現と比較して上昇しました( P <0.05)。 si-NCグループと比較すると、si-PSAT1グループ( P )ではmiR-133a-3pの発現に顕著な変化はありませんでした。> 0.05)。 miR-133a-3p模倣グループと比較して、miR-133a-3p発現は、miR-133a-3p模倣+ oe-PSAT1グループ( P )で有意差を示さなかった。> 0.05)(図7a)。
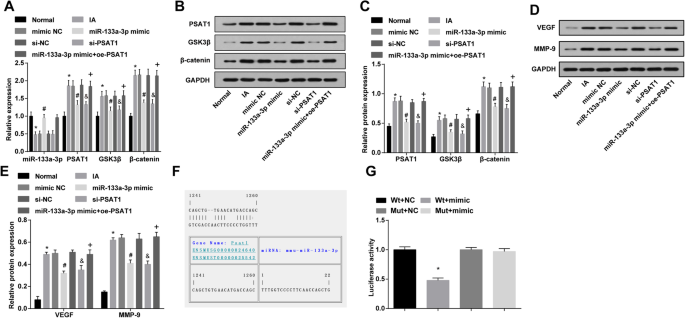
miR-133a-3pの過剰発現とPSAT1の低発現は、in vivoでのIA組織におけるPSAT1、GSK3β、β-カテニン、VEGF、MMP-9の発現を減少させ、PSAT1はmiR-133a-3pの標的遺伝子です。 a RT-qPCRによる各グループのラットのIA組織におけるmiR-133a-3p、PSAT1、GSK3β、およびβ-カテニンの発現の検出。 b PSAT1、GSK3β、およびβ-カテニンのタンパク質バンド。 c ウエスタンブロット分析による各グループのラットのIA組織におけるPSAT1、GSK3β、およびβ-カテニンタンパク質発現の検出。 d VEGFおよびMMP-9のタンパク質バンド。 e ウエスタンブロット分析による各グループのラットのIA組織におけるVEGFおよびMMP-9タンパク質発現の検出。 f ターゲットスキャンによる対応するmiR-133a-3pへのPSAT1結合のターゲットサイトの予測。 g デュアルルシフェラーゼレポーター遺伝子アッセイの結果。 a – e 、 n =12; f – g 、 N =3、* P <0.05vs。通常のグループ/ Wt + NCグループ。 # P <0.05vs。模倣NCグループ。 & P <0.05 vs.si-NCグループ。 + P <0.05 vs.miR-133a-3pミミックグループ。測定データは平均±標準偏差として表され、データは一元配置分散分析とそれに続くテューキーの事後検定によって評価されました
インビボでのIA組織におけるPSAT1、GSK3β、およびβ-カテニンの発現は、ウエスタンブロット分析およびRT-qPCRによって試験された。正常群とは対照的に、IA群のPSAT1、GSK3β、およびβ-カテニンの発現が増加したように見えた(すべての P <0.05)。 IAグループ、模倣NCグループ、およびsi-NCグループにおけるPSAT1、GSK3β、およびβ-カテニンの発現は、著しく変化しませんでした(すべての P > 0.05)。 miR-133a-3pミミックグループおよびsi-PSAT1グループのPSAT1、GSK3β、およびβ-カテニンの発現は、ミミックNCグループおよびsi-NCグループ(すべて P )と比較して減少しました。 <0.05)。 miR-133a-3pミミックグループと比較して、PSAT1、GSK3β、およびβ-カテニンの発現は、miR-133a-3pミミック+ oe-PSAT1グループ(すべて P )で増強されました。 <0.05)(図7a–c)。
ウエスタンブロット分析を使用して、インビボでのIA組織におけるVEGFおよびMMP-9の発現を検証した。結果は、正常群と比較して、IA群のVEGFおよびMMP-9発現が増強された( P の両方)と認識した。 <0.05)。 IAグループ、模倣NCグループ、およびsi-NCグループでのVEGFおよびMMP-9の発現は、明らかに変化しませんでした(すべての P > 0.05)。模倣NCグループおよびsi-NCグループに関連して、miR-133a-3p模倣グループおよびsi-PSAT1グループにおけるVEGFおよびMMP-9の発現は低下しました(すべて P <0.05)。 miR-133a-3pミミックグループと比較して、VEGFおよびMMP-9の発現はmiR-133a-3pミミック+ oe-PSAT1グループで上昇しました(すべて P <0.05)(図7d、e)。
PSAT1はmiR-133a-3pのターゲット遺伝子です
オンライン予測ソフトウェア(https://cm.jefferson.edu/rna22/Precomputed/)を利用して、miR-133a-3pに結合するPSAT1の標的部位、および3'UTR領域の配列を予測および分析しました。 PSAT1およびmiR-133a-3p。 miR-133a-3pの予測される結合部位がルシフェラーゼ活性の変化をもたらすことを証明するために、miR-133a-3p結合部位を削除するPSAT13'UTRの変異配列と野生配列を考案しました。ルシフェラーゼ活性は、血管ECにおけるmiR-133a-3pミミックとWT(Wt-miR-133a-3p / PSAT1)またはMUT(Mut-miR-133a-3p / PSAT1)組換えプラスミドの同時トランスフェクションによって検証されました。その結果、miR-133a-3pミミックはMut-miR-133a-3p / PSAT1グループ( P )のルシフェラーゼ活性に明確な影響を及ぼさないことが明らかになりました。> 0.05)、Wt-miR-133a-3p / PSAT1グループのルシフェラーゼ活性は著しく低下しました( P <0.05)(図7f、g)。
ディスカッション
IAは頭蓋内動脈の異常な拡張であり、血管壁を継続的に外側に押し出すことで動脈壁を弱め、動脈瘤破裂のリスクを高めます[17]。 Liu et al。が実施した研究では、いくつかのmiRNAが血管平滑筋細胞の細胞増殖の調節に関与していることが示されています。これはIAと密接に関連しています[18]。また、最近の研究は、循環miRNAが高リスクの個人で発生したIAの可能性を評価するための新しいバイオマーカーとして使用できるという証拠を提供しました[19]。通常、PSAT1は統合失調症のスペクトラム状態に関与し、セリン代謝を変化させる可能性があると考えられています[20]。現在の研究は、miR-133a-3pが血管内皮損傷を調節し、PSAT1 /GSK3β/β-カテニンシグナル伝達経路を調節することでIAを誘発するという調節的役割を探求するように設計されました。
本研究では、miR-133a-3pの発現とIAの臨床病理学的特徴との関係を分析した結果、miR-133a-3pの発現は年齢、性別、形状、動脈瘤の位置とは関係がないことが示された。動脈瘤の数とサイズに関連付けられています。一部の学者は、動脈壁の局所血流の剪断応力が、血管壁内の線維芽細胞および血管ECによる単球走化性タンパク質-1(MCP-1)およびマクロファージ炎症性タンパク質1α(MIP-1α)発現の誘導を誘導したと考えた。反応性の高い走化性因子MCP-1およびMIP-1αは、血管壁でマクロ食細胞の凝集を起こし、炎症反応を媒介し、核転写因子c-Junの興奮を誘発し、活性化タンパク質1(AP- 1)次に、その構造ドメインでMMP-9プロモーターを活性化して、MMP-9 mRNAの発現を上昇させ、最終的に血管壁の細胞外マトリックスの溶解を誘導し、頭蓋内動脈瘤の形成を引き起こしました[21、22、23]。齋藤ほか[24]は、MMP-9陽性細胞が主に動脈マクロファージの中膜および外膜に由来することを発見し、マクロファージによって発現されるMMP-9が動脈壁の変性を媒介し、動脈瘤の形成につながることを証明した。上記の研究は、MMP-9がIAの形成に関連していることを示しています。私たちの研究の結果は、MMP-9がIAでアップレギュレーションされていることを明らかにしました。したがって、我々は、mR-133a-3pがPSAT1 /GSK3β/β-カテニン経路を調節し、MMP-9をさらに調節することにより、IAの発生と発達に関与しているのではないかと推測した。私たちの研究では、miR-133a-3pを復元すると、頭蓋内動脈瘤組織におけるPSAT1、GSK3β、β-カテニン、およびMMP-9の発現が低下することがわかりました。今後の調査で関連する調査を実施し、調査結果を検証します。
私たちの研究は、miR-133a-3pの発現が減少し、PSAT1、GSK3β、およびβ-カテニンがIAで上昇したという概念に関連する実質的な証拠を提供しました。新たな証拠は、miR-133a-3pがさまざまな種類の腫瘍で抑制的な役割を果たすことを示唆しています。最近の研究では、miR-133a-3pの発現は、非癌組織とは対照的に、乳癌組織で劇的に低下したことが示されています[25]。別の研究では、miR-133a-3pの発現は、隣接する正常組織または良性前立腺病変組織、特に骨転移性PCa組織と比較して、進行性前立腺癌(PCa)組織で低下しているとされています[26]。他のタイプの疾患におけるPSAT1の促進効果は、いくつかの文献に見られます。非小細胞肺癌(NSCLC)ではPSAT1の発現が著しく上昇し、NSCLC患者の臨床転帰不良が予測されたと報告されています[27]。さらに、PSAT1は、CRC腫瘍で最も高いアップレギュレーション遺伝子であると考えられているだけでなく、化学療法抵抗性疾患の患者でも高度に発現しています[28]。 GSK3β活性が癌性組織で上昇したことが明らかにされています[29]。さらに、GSK3βのリン酸化レベルと核のβ-カテニンの発現も増強されており、GSK3β/β-カテニン経路がオステオポンチンの調節に関与している可能性があることを示唆しています[30]。
miR-133a-3pのアップレギュレーションとPSAT1のダウンレギュレーションは、アポトーシスを抑制し、IA ECの増殖と移動を促進し、VEGFを減少させ、IA組織におけるMMP-9の発現を減少させることを示唆するデータから他の結果が明らかになりました。 miR-133a-3pの過剰発現は、I型コラーゲンα1(COL1A1)を標的とすることにより、口腔扁平上皮癌細胞の浸潤、成長、および有糸分裂を再訓練することが以前に示唆されています[31]。高度に発現したmiR-133a-3pは、COL1A1を標的とすることにより、ESCC細胞の増殖を抑制し、細胞アポトーシスを促進し、ESCC細胞の遊走と浸潤を低下させることができると報告されています[32]。別の研究では、miR-133a-3pの一過性のアップレギュレーションが、組換えシグナル結合タンパク質Jκを直接標的とすることにより、胆嚢癌細胞の遊走、浸潤、および増殖能力を抑制することが確認されています[33]。同様に、この研究は、miR-133a-3pがPSAT1を標的とすることによりIAでその役割を果たしていることを示唆しています。 PSAT1の過剰発現はinvitroでESCC細胞の増殖とマトリゲル浸潤を促進し、PSAT1の高発現を伴うESCC細胞をマウスに注射するとinvivoで腫瘍形成が誘導されることが示されています[14]。他の研究でも、PSAT1は高度に発現しており、患者の臨床転帰が不良であると予測されているだけでなく、invivoおよびinvitroで細胞の腫瘍形成と増殖が促進されることが報告されています[13]。以前の研究では、PSAT1が機能喪失および機能獲得実験を通じて細胞周期の進行、増殖、および腫瘍形成を促進することが一般的に確認されています[27]。 MMPは、Zn 2+ を介した活性部位結合水分子の活性化を参照する保存されたメカニズムに基づいてタンパク質基質を切断する一連の酵素で構成されていることが示されています。 イオン[34]。 MMP-9は、多くの生物学的プロセスに計り知れない影響を与える別個のプロテアーゼです[35]。ある研究では、MMP-9は対照群と比較して動脈瘤群で上昇していると主張しています[36]。血管内皮増殖因子-A(VEGF-A)は、栄養膜の血管内分化の重要な調節因子として認識されています[37]。ある研究により、PSAT1の発現が抑制されると、VEGF、β-カテニン、およびGSK3βのリン酸化の発現が抑制されることが明らかになりました[10]。
結論
簡単に結論として、私たちの研究は、miR-133a-3pの過剰発現またはPSAT1のダウンレギュレーションが、IAのGSK3β/β-カテニン経路を阻害することにより、内皮細胞の損傷を抑制し、内皮細胞の増殖を促進するという仮説を確認します。これらの発見は、IAの新しい標的療法における新しい洞察を提供します。これらの発見は、PSAT1 /GSK3β/β-カテニン経路に関連するIAにおけるmiR-133a-3pの役割を強調しています。ただし、miR-133a-3pとPSAT1の効果についての結論は、これに関する既知の研究が限られているため、明確に行うことはできません。厳密に監視し、将来の臨床試験で適切に報告する必要があります。
データと資料の可用性
該当なし
略語
- miR-133a-3p:
-
MicroRNA-133a-3p
- IA:
-
頭蓋内動脈瘤
- PSAT1:
-
ホスホセリンアミノトランスフェラーゼ1
- GSK3:
-
β-グリコーゲンシンターゼキナーゼ3β
- SAH:
-
くも膜下出血
- MiRNA:
-
マイクロRNA
- CRC:
-
結腸直腸がん
- ESCC:
-
食道扁平上皮がん
- HE:
-
ヘマトキシリン-エオシン
- PBS:
-
リン酸緩衝生理食塩水
- MMP:
-
マトリックスメタロプロテアーゼ
- VEGF:
-
血管内皮増殖因子
- NC:
-
ネガティブコントロール
- DAB:
-
ジアミノベンジジン
- FBS:
-
ウシ胎児血清
- UEAI:
-
Ulexeuropaeus凝集素I
- PI:
-
ヨウ化プロピジウム
- FITC:
-
V-フルオレセインイソチオシアネート
- DMSO:
-
ジメチルスルホキシド
- OD:
-
光学密度
- SD:
-
Sprague-Dawley
- SPF:
-
特定病原体除去
- GAPDH:
-
グリセルアルデヒドリン酸デヒドロゲナーゼ
- BCA:
-
ビシンコニン酸
- 3'UTR:
-
3 '非翻訳領域
- WT:
-
野生型
- MUT:
-
ミュータント
- ANOVA:
-
分散分析
- NSCLC:
-
非小細胞肺がん
- VEGF-A:
-
血管内皮増殖因子-A
ナノマテリアル
- エミッタ接地アンプ
- ICAをロードしたmPEG-ICAナノ粒子の調製とLPS誘発性H9c2細胞損傷の治療におけるそれらの応用
- アルテスナートのナノ粒子送達は、ミトコンドリアを介した細胞アポトーシスを活性化することにより、抗腫瘍効率を高めます
- 低温での急速熱アニーリングプロセスによる成長の制御高均一性セレン化インジウム(In2Se3)ナノワイヤ
- 蒸発誘起自己組織化と強化されたガス検知特性によるワームホールのようなメソポーラス酸化スズの容易な合成
- サイズ、修正、欠陥、およびドーピングによるグラフェン仕事関数の設計と調整:第一原理理論研究
- 後部に黒色シリコン層を備えた結晶シリコン太陽電池の調査
- ヒドロキシル基を介したカーボンナノチューブのスライドロール運動モードの調整
- 薄膜シリコン太陽電池用の両面ピラミッド格子を使用した効果的な光吸収
- リトコール酸で修飾された金ナノ粒子の肝臓癌細胞に対するアポトーシス効果
- 小さな希土類フッ化物ナノ粒子は、電気極性相互作用を介して腫瘍細胞の成長を活性化します