


直接還元プロセスの理論的側面
直接還元プロセスの理論的側面
鉄鉱石の直接還元プロセスでは、固体金属鉄(Fe)は、鉱石または金属を溶融させることなく、固体鉄鉱石から直接得られます。直接還元は、酸素(O2)電位での固体の還元として定義できます。これにより、酸化鉄は還元されますが、他の酸化物(MnO、SiO2など)は対応する元素に還元されません。還元は固体状態であるため、これらの元素が還元された鉄に(低い熱力学的活性で)溶解する可能性はほとんどなく、鉄よりも安定している酸化物は本質的に還元されないままです。鉄鉱石の直接還元は、上昇するガスによって高炉のシャフトでも行われます。
鉄–酸素システム
鉄-酸素(Fe-O)システムは、おそらく最も広く研究されているシステムの1つです。システムの熱力学はよく理解されており、酸化鉄が関与するガス状還元の動力学について多くの情報が利用可能です。 Fe-Oシステムで全圧1kg/ sq cmで400℃から1400℃の間で発生する熱力学的に安定した固相を2値図に示します(図1)。この図は、FeがO2とともに、(i)ヘマタイト– Fe2O3、(ii)マグネタイト– Fe3O4、およびウスタイト– FexOの3つの安定した固体化合物を形成することを示しています。ここで、xは1より少し低いです。非化学量論的FeO相(ウスタイト)は570℃以下では不安定で、金属FeとFe3O4の混合物に分解します。したがって、570℃未満の一定温度で相図を右から左に読み取ると、相シーケンスはFe2O3 – Fe3O4 – Feですが、570℃を超えると、シーケンスはFe2O3 – Fe3O4 – FeO –Feになります。
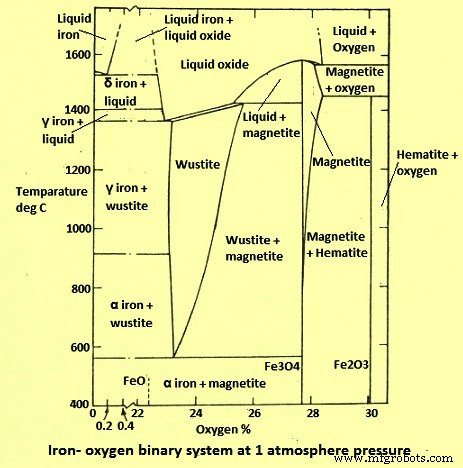
図1Fe-Oバイナリシステム図
固体のアルファおよびガンマ鉄へのO2のごくわずかな溶解度は、O2の0.01%未満です。したがって、O2含有量は、固体のFe修飾の遷移温度に影響を与えず、図では無視されます。
反応平衡を考慮すると、Fe酸化物の還元には、これらのステップの1つ以上が含まれます(i)ヘマタイト(Fe2O3)->マグネタイト(Fe3O4)、(ii)マグネタイト(Fe3O4)->鉄(Fe)、(iii)マグネタイト( Fe3O4)-> wustite(FeO)、および(iv)wustite(FeO)->鉄(Fe)。
ウスタイトは570℃を超える温度でのみ安定です。上記の反応の熱力学的平衡は、使用される2つの主要なガス状還元剤、つまり水素(H2)と一酸化炭素(CO)でよく知られています。
鉄–酸素–炭素システム
COおよびCO2(二酸化炭素)ガスと固体炭素(C)の混合物によるFeおよびFe酸化物の平衡を図2に示します。
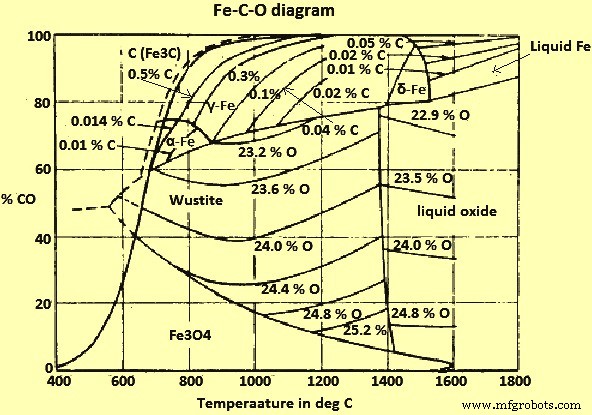
図2Fe-CO-Oシステム図
図2から、710℃を超える温度および1 kg / sq cmの全圧で、すべてのFe酸化物は、Cと平衡状態にあるCO / CO2ガス混合物によって還元され、次のことができると推測できます。したがって、C自体によって削減されます。低温では、Cで過飽和になり、したがってBoudouard平衡に従って、Cの堆積に向かって反応する混合物のみが、ウスタイトに対して還元作用を示します。
鉄–水素–酸素システム
ガスH2とH2O(蒸気)の混合物を含むFeとFe酸化物の平衡状態図を(図3)に示します。
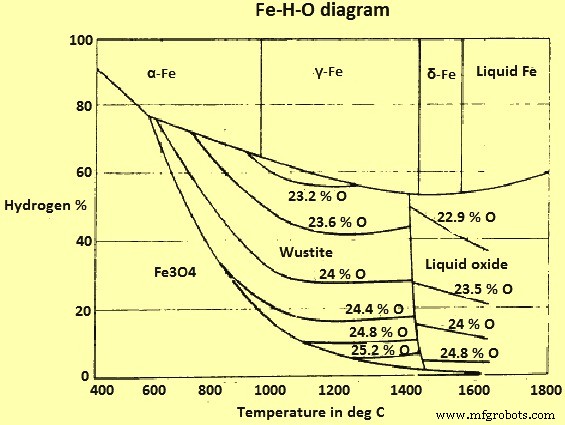
図3Fe-H-Oシステム図
このシステムとFe-O-Cシステムの主な違いは、「スージングライン」または対応する現象がないことです。したがって、理論的には、任意の温度でヘマタイト(およびマグネタイト)をH2でFeに還元することが可能です。
COとH2による削減の比較
Fe-C-OおよびFe-H-Oシステムの研究(図4)から、815℃を超えると、H2はCOよりも効率的な還元剤であるように見えます(つまり、平衡H2/H2O比は対応するCOよりも低くなります/ CO2比)、低温では逆になります。しかし、平衡に近づくにつれて還元速度が非常に遅くなるため、これらの平衡は工業炉ではほとんど達成されません。条件が平衡から明らかに逸脱している場合、H2およびCOによる還元のそれぞれの反応速度は、平衡の考慮から通常予想されるものとは逆の順序になります。したがって、H2は実際には、815℃未満の温度で動作するように設計された非平衡プロセスのより効率的な還元剤であり、COはより高い温度でより効率的です。
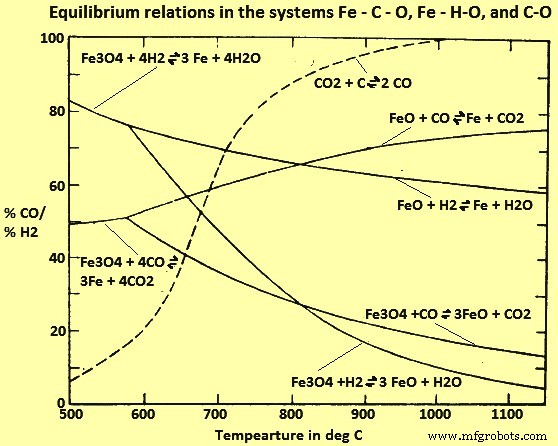
図4Fe-C-O、Fe-H-O、およびC-Oシステムの平衡関係
異なる温度でのCOとH2のガス組成混合物の影響に関する研究は、還元ガス混合物のH2含有量が増加するにつれて、反応速度が増加することを示しています。この関係は著しく非線形であることがわかっています。
Fe酸化物のH2による金属Feへの還元は吸熱性であり、必要な温度を維持するために外部熱源が必要です。 COとの対応する反応は発熱性であり、適切に制御された条件下では、反応は熱的に自立します。実際、チャージの過熱を避けるために、COをH2または他の熱吸収ガスで希釈する必要がある場合があります。一部のプロセスは、CO-H2熱収支を利用し、これらのガスの混合物を使用して、周囲温度から約1100℃の最大反応温度まで鉱石の加熱中に得られる還元量を増やすように設計されています。
図4は、ガス還元が経済的に可能な範囲内のすべての温度で、平衡ガス混合物に少なくとも60%のCOおよび/またはH2が含まれていることを示しています。平衡に達しない場合、これらのガスの未反応濃度はさらに高くなり、大部分はそのまま還元炉を通過します。プロセスが経済的である場合、ウスタイトを金属Feに還元した後に残るガスを、より高次のFe酸化物をウスタイトに還元するため、および/またはガス混合物の再生のために利用する必要がある。ガス状反応生成物の除去。
気固反応と固固反応
気固反応は技術において主要な役割を果たし、鉱石からの金属の抽出(酸化鉄の還元など)を含む非常に幅広い分野を網羅しています。すべての気固反応システムに共通する特徴は、プロセス全体にいくつかの中間ステップが含まれる可能性があることです。典型的には、これらの中間ステップは、(i)気相の大部分から反応する固体粒子の外面への反応物および生成物のガス状拡散(質量移動)、(ii)ガス状反応物またはガス状生成物の細孔を通る拡散を含む。固体反応生成物または部分的に反応した固体の細孔を介して、(iii)固体表面へのガス状反応物の吸着および固体表面からの反応生成物の脱着、および(iv)吸着されたガスと固体との間の実際の化学反応。 / P>
気固反応の分野では、反応の進行と気固反応が行われる炉の性能に影響を与える可能性のある他のいくつかの現象があります。これらの他の現象には、熱伝達、反応に伴う固体構造の変化(焼結など)、および気固反応が行われている炉を通るガスおよび固体の流れが含まれます。削減率は、使用するプロセスに応じて、これらの要因によって制御されます。
固体間の反応は、2つの主要なグループに分けることができます。すなわち、(i)真の固体–互いに接触している2つの粒子間で固体状態で、または固体状態の粒子の移動によって起こる固体反応(例:形成) Fe酸化物とC)の間の反応、および(ii)ガス状中間体を介して起こる固体反応物間の反応(たとえば、1 kg / sq cmの圧力での炭素によるFe酸化物の還元)による炭化鉄の反応。
固体CによるFe酸化物の還元も、非常に低い絶対圧力で実行される場合、真の固体-固体反応である可能性があります。 0.0005 mm Hg(水銀)の真空下で微粉末グラファイト(C)とヘマタイト鉱石の混合物の反応によって行われた研究の1つで、900℃までの温度で反応が非常に起こることがわかりました。ゆっくりと、18時間でFe3O4とFeOのみが形成され、Feは形成されませんでした。試験中、かなりの変化が高温でのみ観察されました。研究中に、反応速度は酸化物相内のFeイオンの拡散によって決定されると結論付けられました。別の調査で行われた推論は、Cが酸化鉄に拡散するということでした。これはおそらく歴史的な関心事にすぎません。しかし、これらの研究は、粉末混合物の上のガス圧が増加すると、反応速度が明らかに増加することを示しています。 N2(窒素)の流れがCとFeの酸化物の混合物を通過した同様のタイプのテストでは、N2の流れが増加するにつれて反応速度の著しい低下が観察されました。これらすべての調査は、真空中またはN2下で行われたかどうかにかかわらず、COまたはH2中の同様の粉末Fe酸化物の還元率で、Cと鉱石の直接的な固体反応(実際のメカニズムと見なされることもあります)を証明しました。真の直接還元の)は、工業炉での還元プロセスの進行には重要ではありません。
還元鉄の細孔構造
いくつかの天然鉱石の還元性試験は、鉄鉱石粒子の多孔性が還元性を制御する最も重要な要因の1つであることを示しています。 90%の還元に必要な時間の逆数として表される還元性は、気孔率によって直接変化しました。 「相対還元性=(多孔性x 0.75)+ 8.0」の式で与えられるように、相対還元性は多孔性の増加とともに増加します。
Fe酸化物の還元は、常に多孔質の反応生成物を生成します。酸化物の性質と還元条件は、還元鉄の細孔の構造に影響を与えます。これは、還元が粒子の表面から内側に向かって進行するためです。元のウスタイト表面によって定義された空間を占める体積が減少します。これは、多孔性を発達させることによってのみ達成することができます。この多孔性の走査型電子顕微鏡写真研究は、一般に、H2還元がCO還元によって得られるものよりも微細な細孔構造を与えることを示した。また、走査型電子顕微鏡写真から、H2の還元温度が600℃から1200℃まで徐々に上昇するにつれて、細孔構造が粗くなることが明らかになります。
酸化鉄の初期細孔表面積は、ガス状還元によって形成される還元鉄の細孔表面積に影響を与えます。酸化鉄の初期細孔表面積が減少すると、還元鉄の細孔表面積が減少します。 BET(Brunauer-Emmett-Teller)技術によって測定されたH2のヘマタイト鉱石から還元された鉄の細孔表面積は、還元温度の上昇とともに減少することを示しています。
還元温度と平均臨界細孔径と最小細孔半径との関係は、細孔径分布から得られました。細孔径は、900℃までの還元温度でゆっくりと増加することがわかっていますが、温度がさらに上昇すると急速に増加します。これらの結果は、走査型電子顕微鏡による破面の観察と一致しており、900℃を超える還元温度での細孔構造の明確な粗大化を示しました。
還元されたヘマタイト鉱石の細孔表面積は、還元温度とガス組成の影響も受けます。 CO / CO2ガス混合物で還元されたヘマタイトから得られる細孔表面積は、H2/H2Oガス混合物で還元されたものの約3分の2です。これは、顕微鏡で観察されたCO/CO2還元鉄の粗い細孔構造と一致しています。
還元鉄の細孔内のガス拡散が測定されました。多孔質媒体内の拡散流束は、2つの拡散プロセス、つまり(i)圧力に依存せず、T(温度)の1/2乗に比例するクヌーセン拡散、および(ii)圧力に反比例してTに比例する分子拡散によって発生します。パワー3/2。
制限的な理想的な構造は、すべてが相互接続され、45度の角度で互いに交差する均一なサイズの細孔を持っていると想定されています。
有効拡散係数は、温度と圧力によって特定の多孔質媒体によって異なり、バイナリガスペアによって異なります。還元温度を下げると、細孔構造が細かくなります。
削減のモード
天然鉄鉱石粒子または焼結ヘマタイトペレットの還元により、生成物層が形成されます。このよく知られた現象は多くの研究の対象でした。 H2による焼結ヘマタイトペレットの還元に関する最近の研究の1つでは、部分的に還元されたヘマタイトペレットの研磨部分に層が形成される典型的な例があることに気づきました。層間の比較的滑らかな界面は通常、低倍率で表示されますが、そのような外観は誤解を招く可能性があります。
これは、ガス拡散がウスタイト層で十分であり、進行するFe/FeO界面の前に内部還元を与えることを示しています。 (i)温度が下がり、(ii)気孔率が上がり、(iii)粒子サイズが小さくなると、内部還元のゾーンが広がります。
所定の割合の減少を達成するために必要な時間に対する粒子サイズの影響は、減少のモードに依存し、したがって、速度制御プロセスのタイプに依存します。ガス状還元による多孔質Fe酸化物の還元モードの考察は、3つの制限速度制御プロセス、すなわち(i)均一な内部還元、(ii)制限混合制御、および(iii)多孔質Fe層での拡散を示しています。還元がこれらのいずれかによってのみ制御される場合、還元の時間は、3つの方法のいずれかで粒子(球状)の直径に関連します。つまり、(i)時間は直径に依存しない均一な内部還元、(ii)混合制御の制限、および(iii)多孔質鉄への拡散。
速度制御プロセスは、(i)均一な内部還元があり、したがって小さな粒子サイズが必要な場合、または(ii)粒子サイズが支配的であるため、鉄層の細孔内のガス拡散による最終的な速度制御が支配的である場合にのみ、比較的単純になります。は大きい。温度、ガス組成、粒子サイズ、および酸化物の種類に応じて、還元が進行するにつれて、ある制限速度制御プロセスから別の制限速度制御プロセスへの移行があり得ることもまた認識されるべきである。酸化鉄の還元は、説明のつかない異常な行動を示すこともあります。
多孔質鉄鉱石粒子の還元率
鉱石の多孔性と細孔構造は、内部還元の程度と均一性に顕著な影響を及ぼします。研究の1つでは、90%COと10%CO2の混合物、および1000℃のH2のヘマタイト鉱石の還元速度に対する粒子サイズの影響は、粒子サイズの増加に伴って内部還元が制限されることを示しています。粒子の外側の領域に移動するため、粒子サイズが大きくなると全体的な減少率が低下します。
多孔質ヘマタイト粒子の還元の初期段階では、FeOへの急速な変換とそれに続くFeOからFeへの内部還元があります。酸化鉄の細孔内でのほぼ完全なガス拡散の限定的な場合では、内部還元が支配的であり、速度は主に細孔壁での気固反応によって制御されます。数原子の厚さのFe層は、FeOの細孔壁を覆うと想定されています。還元速度は、細孔壁のFe層のコーティングを介したO2の急速な拡散と、この非常に薄いFe層の表面のO2とのH2またはCOの化学反応によって共同で制御されると推定されます。
>粒子サイズの影響は、粒子のサイズが小さくなると減少率が増加することを示しています。典型的な顕微鏡写真は、還元のモードが粒子内で粒子ごとに異なることを示しています。これは、酸化物粒子の多孔性の局所的な違いによるものです。細孔径のばらつきと大きな細孔でのガス拡散の高速化により、ほとんどの反応は大きな細孔の壁で発生します。つまり、全細孔表面積のごく一部のみが反応に使用されると予想されます。さまざまなタイプのヘマタイト鉱石粒子で達成される800℃でのH2還元率は、形成されるFe(またはFeO)の細孔表面積の増加とともに非線形に増加します。これらの結果は、細孔表面積が大きいほど、反応に使用される細孔壁全体の割合が小さいという事実を実証しています。
H2 – COガス混合物の内部還元率は、通常、H2とCOによる2つの個別の還元率の合計です。還元データと観察されたC堆積の両方は、1000℃未満でガス反応が水につながることを示しています。ガスの平衡が遅い。
鉄鉱石(塊またはペレット)の還元率
塊状鉱石または鉱石ペレットの還元速度は、充填床内の還元ガスの流れにおいて複雑な性質を持っています。複雑さは、還元の全体的な速度が、熱や質量などの一連のいくつかの反応プロセスによって制御されるためです。ガス膜境界層を介した移動、多孔質生成物層における気固反応およびガス拡散。コンピューター計算によって促進される数学的分析を通じて、さまざまな還元モードでの大きな酸化物粒子の還元速度を説明するために、多数の方程式が導き出されました。
単一のペレットまたは鉄鉱石粒子を使用したいくつかの実験では、熱伝達は比較的速く、十分に高速のガス流では、ガス膜の物質移動抵抗は無視できるほど小さい。したがって、還元速度に影響を与える直列の主に2つの主要な反応ステップ、すなわち(i)ガス酸化物反応、および(ii)多孔質酸化物および多孔質生成物層でのガス拡散があります。これらの速度プロセスの相対的な影響は、粒子サイズ、ガス組成、温度、および還元モードに依存し、還元の進行とともに変化します。
多孔質Fe層でのガス拡散
研究の1つでは、Fe層の細孔内でのガス拡散の影響を実証するために、一方向の還元実験が実施されました。長いシリンダーのサンプルは、塊状のヘマタイト鉱石の大きな断片から準備され、ぴったりと合うニッケル管の中に詰められました。必要な時間H2を減らした後、サンプルを軸方向に分割して研磨し、Fe層の厚さを測定しました。実験の結果、還元されたFe層の厚さが約1mmの場合、細孔拡散律速の結果と同様に、放物線状の反応速度式と一致してさらなる還元が進行することが示された。これらのテストは、多孔質のFe層の厚さが増すにつれて、還元速度が最終的にFe層の細孔内のガス拡散によって制御されることを示しています。
Fe層の主な前進前線に先行する部分的な内部還元は、還元された層にいくらかのFeOを閉じ込めることにつながる可能性がある。この状況は、還元の最終段階でのO2の除去が遅くなる可能性があります。
還元温度が下がると、細孔構造ははるかに細かくなり、おそらく、接続されたキャピラリーに多くの狭いチャネルとボトルネックがあり、クヌーセン拡散が支配的であるため、有効分子拡散係数/有効平均クヌーセン拡散係数の比率の値が低くなります。還元温度を上げると細孔構造が粗くなり、ガスが細孔を通過しやすくなるため、比率が高くなります。
H2-CO-CO2混合物(CO / CO2比)によって900℃で還元された焼結ヘマタイト鉱石ペレットおよびマグネタイト鉱石ペレットの50%、75%、90%、および95%の還元を達成する時間に対するガス組成の影響煤の堆積を抑制するために9に等しい)、H2がCOに置き換えられると、O2除去の所定の割合を達成するための等温還元の時間は、約50%COまで徐々に増加し、COをさらに追加すると、削減の時間。 100%(CO / CO2比が9に等しい)の還元時間は、同じ温度でのH2の約10倍です。ガスの運動論から導き出された、H2-H2OやCO-CO2などのバイナリシステムにおける分子ガス拡散率は、システムに対して不変であり、本質的にガス組成に依存しません。ただし、3成分系および多成分系では、各種の拡散係数が異なり、ガス組成によって異なります。さらに、拡散流束の反応速度式は複雑です。
H2-CO混合物中のヘマタイト鉱石ペレットの還元挙動は、H2およびCOで観察されたものと同様のパターンを示しています。これは、約50%を超える還元速度がFeの細孔内のガス拡散によって制御されることです。レイヤー。
初期レートでの混合制御の制限
還元の初期段階では、還元速度は、(i)FeOの細孔内でのガス拡散(FeOでの固体拡散は無視できます)、および(ii)FeOの細孔壁での反応によって共同で制御されます。 。これは、薄い多孔質のFe層とその中の急速なガス拡散を意味します。 FeOの多孔性とその中のガス拡散係数に応じて、公称Fe/FeO界面の前に部分的な内部還元があります。 H2と多孔質FeOの反応は、通常、公称Fe/FeO界面に近い細孔口に限定されます。
部分的な内部削減
ガス組成、温度、ペレットサイズ、および総ガス圧に応じて、制限速度法の枠内で、削減のある期間中に混合速度制御が行われます。速度方程式は通常、ペレットのガス状還元が、ペレットの粒子間細孔を通るガスのゆっくりとした向流拡散と、ガスとFe酸化物との遅い化学反応によって共同で制御されるという仮定に基づいています。粒子のFe酸化物-/Fe界面。
水性ガスシフト反応
水性ガスシフト反応は、酸化鉄の還元における還元剤として改質された炭化水素を使用する直接還元プロセスにおいて重要な役割を果たします。 COとH2による鉄鉱石の還元速度の違いと、CO / CO2混合物に含まれる少量のH2でも還元速度に及ぼす顕著な影響から、H2が実際の還元成分であることが一般的に認められています。そのようなガス混合物で。 COは、主に、結果として生じる蒸気(H2O)をH2に戻すために役立つと考えられています。反応は、(i)H2 + FeO =H2O + Fe、および(ii)H2O + CO =H2+CO2です。
この反応の2番目のサブプロセスは、水性ガスシフト反応として知られています。このプロセスには触媒が必要であることはよく知られています。鉄鉱石の還元では、すべての生成物(Fe3O4、FeO、およびFe)が可能な触媒として考慮されます。これらの中で特に活性なのは固体のFeです。したがって、H2を含むCO / CO2混合物中の鉄鉱石の還元プロセスは、金属Feが存在する場合、反応シーケンスとして理解されます。副反応(i)は適切な還元がFe酸化物表面で行われ、副反応(ii)は水性ガス反応によるH2の再生がFe表面で行われます。
2つのサブ反応の空間的分離には、反応の参加者の1人によるガス拡散または表面拡散として行われる輸送プロセスによる接続が必要です。最適な条件は、Fe/Fe酸化物/ガスの3相境界で発生します。
縮小中の腫れ
鉄鉱石またはペレットの見かけの量は、通常、還元中に増加します。これは腫れと呼ばれます。大きく3種類の腫れ挙動が見られます。これらは、(i)通常の膨潤、(ii)FeOからFeへの変換に伴う突然の体積膨張、繊維状Feのウィスカーワイヤーとして知られるフィラメント状の成長の形で現れるFe、および(iii)破裂膨張、少量のアルカリを含むFeに富む材料の典型的な挙動。この後者の種類の動作は、膨張の大部分が反応生成物としてFeが現れる前に発生するという点で、壊滅的な膨張とは異なります(ただし、それほど深刻ではありません)。
塊鉱石も焼結鉱も異常にまたは壊滅的に膨潤することは知られていないが、特定の種類のペレットは膨潤し、異常に膨潤したペレットは柔らかく、スポンジ状で崩壊する傾向があるため、負担の透過性を低下させることによって操作上の問題を引き起こすと言うことができます。
文献で報告されているさまざまな酸化鉄およびFeの比体積は、Fe 1グラムあたり0.272ccのFe2O3(室温)、Fe1グラムあたり0.270ccのFe3O4、Fe1グラムあたり0.231ccのFeO(23.5%O2)です。 、およびFe1グラムあたり0.128ccのFe。したがって、ボリュームは、削減の各段階で減少すると予想されます。しかし、Fe鉱石の膨潤の主な原因は、六角形のヘマタイト鉱石が立方晶のマグネタイト鉱石に変化し、その結果生じる格子の乱れによって引き起こされます。格子擾乱は細孔形成を引き起こし、それによってヘマタイトからマグネタイトへの変換中にFe鉱石の見かけの体積がかなり増加します。
一般に、COに富むガスの還元中、膨潤はH2に富むガスよりもはるかに大きくなります。この動作の理由は、CO含有ガス混合物でのC堆積中に金属ダスティングが発生するためです。ただし、Cの堆積がない場合、CO-CO2ガス混合物の還元中に発生する可能性のある膨潤を説明することは困難です。減少に伴う腫れや収縮の原因と影響はまだ解決されていません。
通常、鉱石ペレットには2種類の不純物が含まれています。これらは、(i)腫れを妨げる効果のある不純物、および(ii)腫れを改善する効果のある不純物です。最初の例はシリカ(SiO2)で、2番目の例はアルカリ(K2O、Na2O)です。 5%までのSiO2を含む試薬グレードのFe2O3ペレットは、CO-CO2ガス混合物で還元しても膨潤しないことがわかっています。また、強度を維持し、壊滅的な膨潤を防ぐために、酸性ペレットには一定量のSiO2が必要です。 2番目のケースでは、アルカリNa2CO3またはK2CO3を0.1%から1%の範囲で少量添加すると、通常の鉱石ペレットのH2またはCOに壊滅的な膨潤が生じる可能性があることがわかります。アルカリの影響は、ペレットの塩基度(CaO / SiO2)比が高くなるにつれてより顕著になります。細粒の酸性脈石を添加して安定したアルカリケイ酸塩を形成することにより、悪影響を防ぐことができます。
鉱石ペレット中の不純物の影響(石灰含有量など)については、いくつかの矛盾した観察結果があります。ヘマタイト鉱石ペレットへの少量のCaO添加(0.1%未満)は還元中にかなりの膨潤を引き起こし、これはCaOが壊滅的な膨潤の原因であることを示唆しました。一方、ヘマタイト鉱石ペレットに約1%のCaOを添加すると、還元中の膨潤が抑制されることがわかっています。膨潤に対するCaOの観察された効果のこれらの変動は、アルカリなどの鉄鉱石中の他の不純物の有無に起因する可能性があります。
Cによるヘマタイト鉱石の還元
ヘマタイト鉱石とCの間の反応は、金属化鉱石ペレットの調製において基本的に重要です。新しい関心の多くは、直接還元鉄(DRI)の製造で還元剤として固体Cを使用するロータリーキルンプロセスの開発によって刺激されました。 Cによる酸化鉄の還元は、真の固固反応が主なメカニズムである非常に高い真空下を除いて、ガス状中間体COおよびCO2を介して起こると一般に認められています。
Cによるヘマタイト鉱石の還元中に起こるガス状中間体を介した反応メカニズムは、反応(i)C(s)+ 0.5 O2 =CO(g)、(ii)FexOy(s)+ CO(g)=FexOを介して行われます。 (y-1)(s)+ CO2(g)、および(iii)CO2(g)+ C(s)=2CO(g)。
COの初期形成は、全体的な反応速度の重要なステップです。閉じ込められた空気のO2は、Fe酸化物の解離によって放出されたO2ガスと一緒に、Cと反応してCOを生成します(最初の反応)。さらに、一部のCOは、CとFeの酸化物粒子間の接触点で発生する真の直接還元によっても形成される可能性があります。このようにして生成されたCOガスは、ヘマタイト鉱石粒子と容易に反応します(2回目の反応)。 BoudouardまたはCO2ガスとC粒子間の溶液損失反応は、COガスを再生し(3番目の反応)、それによってサンプルの細孔内に含まれる気相の還元電位を回復する傾向があります。 CO2中の特定の種類のCの酸化は、特定の金属および金属化合物の存在下で触媒されます。 Li2O(酸化リチウム)の添加によりプロセスの速度向上が観察され、FeS(硫化鉄)の添加により抑制効果が報告されています。金属Feは、グラファイト(C)のガス化に適した触媒であることがわかっています。混合物におけるこの予測不可能な触媒反応のため、反応の全体的な速度を説明するための数学的モデリングによって導き出された方程式は、価値が限られており、反応が触媒されないシステムにのみ適用できます。
適度に高い温度(たとえば1000℃)では、Fe酸化物反応の速度(570℃を超える温度で、シーケンスはFe2O3、Fe3O4、FeO、Fe)は、Boudouard反応の速度よりもはるかに大きくなります。言い換えれば、全体的なプロセスは、Boudouard反応によるCOガスの利用可能性によって制限されます。 Thus at steady-state the composition of this gas-phase closely corresponds to the equilibrium gas-phase composition for FexOy/FexO(y-1).
Fe oxides reduction with hydrocarbons
Hydrocarbons can be used in two ways as a reducing agent for the production of DRI. These are (i) direct use of hydro-carbons or a mixture of gas containing hydro-carbons, and (ii) use of the reformed hydrocarbon products (CO, H2), by reforming within the reduction reactor (it has been found that auto-catalytic reforming of some hydro-carbons within the reducing furnace provided an access of macro and micro porosity which leads to more extensive reduction and also which leads to the deletion of the capital cost of gas reformer and processing.
There are a few studies using directly hydrocarbons or a mixture of gas containing hydrocarbons as reductant for direct reduction of iron ores. Two important points emerge from these studies. The first is that the rate of reduction with hydrocarbons is slow and the production of a high quality of DRI is troublesome and uneconomical. The second point is that these studies have been done under isothermal conditions in a thermo-gravimeter with single particle or powder compact, thus the results are of only theoretical value.
Theoretical importance of investigations with hydrocarbons – The kinetics of ferric oxide reduction by pure methane (CH4) has been studied in the three temperature ranges of (i) low temperature (500 deg C to 600 deg C), (ii) medium temperature (650 deg C to 750 deg C) and (iii) high temperature (800 deg C to 950 deg C). At the low temperature, the reduction proceeds only from Fe2O3 to Fe3O4. A prolonged holding of the sample in a stream of CH4 has not led to any process extension beyond this stage. The rate became appreciable at 650 deg C. In special experiments after the Fe3O4 composition has been reached, the sample has been reduced further by H2 and CH4. It has been shown that CH4 reduction in the low temperature range beyond the Fe3O4 stage occurs only if a sufficient quantity of metallic Fe has been built up. In this case the reducing agent has not been CH4, but its decomposition product, H2. C formed by CH4 decomposition takes almost no part in the reduction and gets accumulated in the sample.
In the medium temperature range the conversion of Fe3O4 to FeO takes place but at low rates. A sharp rise in reduction rate is observed on going from 750 deg C to 800 deg C. The process becomes very sensitive to temperature changes beyond 800 deg C, and accelerated considerably in the high temperature range, when metallic Fe appeared in the sample. The appearance of metallic Fe at the FeO to Fe stage, at comparatively high temperatures indicates a decisive role of metallic Fe as a catalyst for reforming CH4 by the reduction products (CO2, and H2O). In the absence of a catalyst, the decomposition of CH4 and its reforming by the reduction products (CO2, H2O) do not occur to any substantial extent and no C accumulation in the sample has been observed. When the Fe catalyst is present, CH4 dissociation into the elements takes place only at very late stages of reduction, when there is insufficient CO2 and water vapour to convert all the CH4 diffused into the sample. C build-up in the sample starts from that stage.
In the 2-stage production of DRI with CH4, it has been found that the complete decomposition of CH4 in the presence of the Fe bearing material occurs at temperatures of 850 deg C to 900 deg C, which is 400 deg C to 450 deg C lower than on an inert surface (e.g. fire clay), while the reaction rate, conversely, has been 10 times higher. The products of the first stage are a sooty Fe containing 30 % to 50 % C and technically pure H2.
In the second stage, the product of the first stage (sooty Fe with highly dispersed C in the pores of DRI and on the surface of the Fe particles) has been used as an active reducing agent and mixed with mill scale or concentrate. The mixture has been reduced in the temperature range 1050 deg C to 1100 deg C with a make-up reducing agent of H2 reformed natural gas. The results of industrial trials has shown that the use of sooty Fe instead of soot, petroleum coke and the other known carbonaceous reducing agents considerably intensified the Fe-oxide reduction process. As is well known, the direct reduction of Fe oxides with C is directly related to the rate of reaction between the C and CO2. The sooty Fe can have intensified the rate of Boudouard reaction.
The isothermal reduction of hematite ore pellets (with 10 % to 15 % porosity) in a thermo-balance with a mixture of CH4-H2 (containing 4.5 % CH4) within the temperature range 700 deg C to 1000 deg C has shown that the reduction is chemical – controlled initially and diffusion – controlled in the later stages. It has been shown that reduction in pure H2 is faster than in the CH4- H2 mixture. This difference is attributed to C deposition in the outer reduced layers of the pellet, causing resistance to gas diffusion when the reducing gas contained CH4. It has been shown that the excess residual C can be removed from the reduced iron at lower temperature by its hydrogenation.
In another study, it has also been demonstrated that it is possible to hydrogenate residual C in direct reduced products to CH4. The C formed as a result of the reduction of Fe oxide in a mixture of CH4 and H2 (containing 20 % CH4) reacted with steam (H2O) according to the water gas reaction to regenerate H2 and produce CO.
Pure ferric oxide briquettes were reduced at temperatures ranging from 800 deg C to 1050 deg C, in gas mixtures containing H2, CO, CH4, N2 and CO2, which has been obtained by partial oxidation of natural gas with air. The CH4 content of the reformed gas mixture was between 13 % and 16 %. The overall reduction rate again has been controlled initially by chemical reaction and the gaseous diffusion has been applicable during the latter stages. It has been shown that the hematite ore briquettes have swelled and considerable porosity has been was developed during reduction. The solid-state diffusion rates increased more rapidly with temperature than it did by interfacial or gaseous diffusion reaction rates. The reduction of porous (30 % porosity) Fe ore in CH4 has indicated that the reaction proceeded stepwise from Fe2O3 to Fe3O4, FeO and Fe. The Fe catalyzed the CH4 cracking reaction. Optimum conditions for CH4 utilization occurred at around 1000 deg C.
The above findings are not consistent with the earlier studies on the understanding of high-grade porous (around 30 %) or dense hematite ore reduction kinetics, which had shown that the rate of reduction can be considered to fall between 3 limiting cases, namely (i) uniform internal reduction, (ii) limiting mixed control, and (iii) diffusion in porous iron layer, respectively with the rate of reduction corresponding to, (i) chemical control, (ii) the overall chemical control and diffusion control, and (iii) diffusion control. The overall rate of reduction is not controlled by only one of these rate controlling mechanisms and can be changed from one limiting case to another during the course of reduction.
In one of the studies it has been found that the most important factors controlling the extent of reduction are (i) the temperature, (ii) the composition of gas, presence of unreacted hydrocarbons in the reducing gas, the ratio of H2/C in it, and reducing capacity, (iii) the ore particle size, and (iv) the residence time for reduction.
Reduction of Fe oxides with the products of CH4 reformed with H, O within the reduction furnace – In early 1981 a commercial process has been introduced, using gaseous mixtures containing upto around 30 % by volume of CH4 (e.g. coke oven gas), for the direct gaseous reduction of Fe ore in a counter current moving bed shaft furnace. The furnace contained a reduction zone, a cooling zone, and an intermediate reforming zone. A hot mixture of coke oven gas and steam has been fed to the intermediate zone and reduced Fe ore therein catalyzed the reforming of the CH4 to CO and H2. The reformed gas flows upward into the reduction zone for the reduction of Fe ore.
製造プロセス