


M2マクロファージ由来のマイクロRNA-18aはTGFBR3を阻害し、TGF-βシグナル伝達経路を介して鼻咽頭癌の進行と腫瘍増殖を促進します
要約
目的
鼻咽頭癌(NPC)は、高い転移性と浸潤性を伴う鼻咽頭疾患の一種です。腫瘍関連の代替活性化(M2)マクロファージは、NPCと接続することが証明されています。これに基づいて、この研究は、NPCのM2マクロファージからのマイクロRNA-18a(miR-18a)のメカニズムと関与を調査することを目的としています。
メソッド
末梢血単核細胞はマクロファージに分化し、マクロファージはインターロイキン-4によってM2型に極性化されました。 SUNE-1およびCNE2細胞に、復元または枯渇したmiR-18aまたはトランスフォーミング成長因子-ベータIII受容体(TGFBR3)をトランスフェクトして、TGF-βシグナル伝達経路の関与を伴うNPC進行におけるそれらの役割を調べました。次に、SUNE-1およびCNE2細胞を、回復または枯渇したmiR-18aまたはTGFBR3で処理したM2マクロファージと共培養し、TGF-βシグナル伝達経路の関与を伴うNPCでのそれらの複合的な役割を理解しました。
結果
MiR-18aは高度に発現し、TGFBR3はNPC細胞で低発現していました。 MiR-18aの回復、TGFBR3ノックダウン、またはmiR-18a模倣体との共培養、またはsi-TGFBR3をトランスフェクトしたM2マクロファージは、SUNE-1細胞の進行、マウスの腫瘍増殖、p-Smad1 / t-Smad1の減少、p-の上昇を促進しました。 Smad3 / t-Smad3。 miR-18aのダウンレギュレーション、TGFBR3の過剰発現、またはmiR-18a阻害剤またはOE-TGFBR3をトランスフェクトしたM2マクロファージとの共培養は、CNE2細胞の進行、マウスの腫瘍増殖、p-Smad1 / t-Smad1の増加、p-Smad3 /の減少を抑制しました。 t-Smad3。
結論
私たちの研究は、M2マクロファージからのmiR-18aが、Smad1の不活性化とSmad3の活性化に加えて、TGFBR3抑制を介してヌードマウスのNPC細胞の進行と腫瘍の成長を促進することを明らかにしています。
はじめに
鼻咽頭癌(NPC)は、局所浸潤および早期遠隔転移に傾く上皮性悪性腫瘍です[1]。 NPC患者はしばしば外転神経麻痺とホルネル症候群を訴えます[2]。現在、適用される治療は主に放射線療法、および統合された放射線療法と化学療法で構成されています[3]。残念ながら、放射線療法と化学療法は予期せぬ合併症を伴い、放射線療法に対する後天的な耐性はNPCの結果を妨げます[3]。潜在的な標的療法を掘り下げるタスクが優先事項であることを考えると。
調節不全のマイクロRNA(miRNA)は、NPCの腫瘍形成、転移、浸潤、および放射線療法と化学療法への耐性に関与していることが報告されています[1]。 miRNAのサブファミリーとして、miR-18aは生殖器1の阻害とmTOR経路の活性化における形態形成の抑制を介してNPCの進行を促進することがわかっています[4]。それに加えて、miR-18aはDICER1調節を介してNPC細胞の増殖と転移を活性化することがさらに検証されています[5]。さらに、NPC細胞の進行は、miRNA生合成障害を介してmiR-18aによって促進されることが証明されています[6]。さらに、miR-18aはNPCの転移において決定的に機能することが報告されています[7]。代替活性化(M2)マクロファージは、固形および血液悪性腫瘍の重要な構成要素であり、進行、転移、および治療抵抗性と関連しています[8]。 M2分極腫瘍関連マクロファージはNPCの予後不良と関連しています[9]。エプスタインバーウイルス陰性とエプスタインバーウイルス陽性のNPCにおけるM2マクロファージの違いが興味深いことに記録されています[10]。 miR-18aがM1マクロファージを誘導することによって結腸癌細胞の肝転移を阻害することを概説する研究があります[11]。トランスフォーミング増殖因子ベータIII受容体(TGFBR3)は、細胞表面のシグナル伝達とバランスを刺激するII型TGF-β受容体リガンドを提供するTGF-β補助受容体であり、可溶性TGFBR3は癌進行中の調節因子です[12]。低発現のTGFBR3は、免疫寛容な腫瘍微小環境を誘発すると報告されています[13]。反対に、TGFBR3の一過性の過剰発現はヒトNPC細胞にアポトーシスを誘導します[14]。私たちの知る限り、miR-181aによって誘導されるM2マクロファージの極性化は、Kruppel様因子6およびCCAAT /エンハンサー結合タンパク質α軸を介してM2マクロファージを介した腫瘍細胞の転移を促進します[15]。
まとめると、多くの研究がNPCにおけるmiR-18a、TGFBR3、およびM2マクロファージの独立した役割を発見しましたが、これら3つの要因間の複合的な相互作用はまだとらえどころのないものです。この研究は、NPCにおけるこれらの要因のメカニズムと参加を解読するために開始されたことを考慮します。
材料と方法
倫理声明
この実験は、中南大学中南大学第3病院の倫理委員会によって承認され、医療倫理基準を満たしていました。この研究は、すべてのドナーからの書面による同意を得て実施されました。動物実験は、国立研究所の動物管理および使用規則の要件に準拠していました。
末梢血単核細胞の収集
単球は、健康なドナーの末梢血から付着法によって得られた。末梢血検体は、中南大学第3中南大学血液学部の健康なドナーから入手しました。単球由来のマクロファージは、健康なドナーからの血液のバフィーコート調製物から密度勾配遠心分離(Ficoll-Paque、GE Healthcare)によって以前に分離された末梢血単核細胞(PBMC)のプラスチック接着によって得られました。次に、2.0×10 6 PBMCは、10%ヒト血清(Millipore、ベッドフォード、マサチューセッツ、米国)およびペニシリン/ストレプトマイシンを添加したダルベッコ改変イーグル培地(DMEM)で、12ウェルプレート(NalgeNunc、ニューヨーク、米国)で培養しました。 PBMCを壁に2〜3時間付着させた後、上澄みと懸濁したPBMCを繰り返し除去して、付着した単球を得ました。
マクロファージ極性化
単球は、ヒトマクロファージコロニー刺激因子(M-CSF)によってマクロファージに分化するように誘導され、インターロイキン(IL)-4によってM2マクロファージに極性化されました。
マクロファージの誘導
単球を20%ウシ胎児血清(FBS)-DMEMで培養し、M-CSF(100μg、Peprotech、ニュージャージー州、米国)を最終濃度100 ng / mLになるように添加しました。培地は3日ごとに半分に更新され、100 ng / mLM-CSFが補充されました。 7〜8日目に培養し、細胞の一部を回収し、マクロファージ表面マーカーCD68 [16]、CD163 [17]、およびCD206 [18]を蛍光抗体法[19]でテストして、マクロファージを同定しました。
マクロファージの極性化
20 ng / mLのIL-4(Peprotech)を分化培地にさらに24時間補充することにより、マクロファージをM2マクロファージに極性化しました。 M2マクロファージサンプルの一部をフローサイトメトリー検出に利用しました。サンプルは3つのチューブに分けられました。チューブ1は、IL-4刺激のない付着性マクロファージの同じバッチでした。チューブ2と3は、IL-4によって刺激された付着性マクロファージでした。ローディング時にサンプルを約10,000細胞に希釈し、非特異的免疫グロブリンGを添加してFc受容体をブロックしました。次に、サンプルに非特異的アイソタイプ抗体、PE標識CD68抗体、およびPE標識CD163抗体(両方とも米国カリフォルニア州バイオレジェンド)を添加しました。 30分間インキュベートし、リン酸緩衝生理食塩水(PBS)中の0.5%ウシ血清アルブミンですすいだ後、サンプルを遠心分離し、500μLのPBSで細胞懸濁液にして検出しました。
逆転写定量的ポリメラーゼ連鎖反応
逆転写定量的ポリメラーゼ連鎖反応(RT-qPCR)を使用して、miR-18a、CCL22、ペルオキシソーム増殖因子活性化受容体γ(PPAR-γ)、および収集した細胞におけるTGFBR3mRNAの発現を検出しました。
全RNAはTrizol(Invitrogen、Carlsbad、CA、USA)によって細胞から抽出され、miR-18aおよびPrimeScriptTMRT用のMir-XmiRNA First Strand Synthesis Kit(Clontech、Mountain View、CA、USA)によって相補的RNAに逆転写されました。 CCL22、PPAR-γ、およびTGFBR3用のマスターミックスキット(タカラ、ダリアン、中国)。 U6およびグリセルアルデヒド-3-リン酸デヒドロゲナーゼ(GAPDH)は、miR-18a、CCL22、PPAR-γ、およびTGFBR3のコントロールをロードしていました。 SYBR @ LightCycler 480 IIシステム(Roche Diagnostics、Indiana、USA)のPremix ExTaq™II(Perfect Real Time)(Takara)をPCRに使用しました。データ計算は2 -△△CT で評価した。 方法。 PCRプライマーを表1に示しました。
<図>ウエスタンブロットアッセイ
ウエスタンブロットアッセイは、収集された細胞内のTGFBR3、総(t)-Smad1、リン酸化(p)-Smad1、t-Smad3、およびp-Smad3タンパク質の検出に適用されました。
細胞の総タンパク質を抽出し、ビシンコニン酸キットに基づいてタンパク質濃度を決定しました。タンパク質サンプルをドデシル硫酸ナトリウムポリアクリルアミドゲル電気泳動でウェルにロードし、ポリフッ化ビニリデン(PVDF)メンブレンに転写しました。 PVDFメンブレンをスキムミルクでブロックし、一次抗体TGFBR3(1:2000、R&D Systems、ミネアポリス、MN、米国)、t-Smad1(1:1000)、p-Smad1(1:1000、Santa Cruz Biotechnology)とインキュベートしました。 、t-Smad3(1:1000)、p-Smad3(1:1000)、およびGAPDH(1:1000、すべてAbcam、ケンブリッジ、マサチューセッツ州、英国)に続いて、西洋ワサビペルオキシダーゼ標識二次抗体( 1:500、Jackson ImmunoResearch Laboratories、PA、USA)。 Tween 20を含むトリス緩衝生理食塩水で3回洗浄すると、化学発光が増強されてメンブレンが発色しました。信号の定量化は、国立衛生研究所のImageJImagingによって完了されました。信号強度がGAPDHに正規化された処理分析ソフトウェア。
細胞培養とスクリーニング
ヒトNPC細胞株CNE2、TW03、C666-1、SUNE-1および正常なヒト鼻咽頭細胞株NP96(上海生物科学研究所、中国科学アカデミー、上海、中国)をロズウェルパーク記念研究所(RPMI)で培養しました- FBS(Gibco)、100μg/ mLペニシリンおよび100μg/ mLストレプトマイシンを含み、80%コンフルエンスで継代した1640培地(Gibco、CA、USA)。 RT-qPCRを利用してmiR-18aの発現を検出しました。これらのNPC細胞株の中で、CNE2およびSUNE-1細胞は、NP96細胞とのmiR-18a発現に最大および最小の差を示したため、miR-18aのダウンレギュレーションまたはアップレギュレーションアッセイに選択されました。
セルのグループ化と処理
すべてのNPC細胞株の中から、miR-18aの発現においてNP96細胞との差が最も小さいSUNE-1細胞を選択しました。 Lipofectamine 2000(Invitrogen)の仕様に基づいて、SUNE-1細胞にmiR-18aミミック、miR-18aミミックネガティブコントロール(NC)、si-TGFBR3、またはsi-TGFBR3NCをトランスフェクトしました。
すべてのNPC細胞株の中で、miR-18a発現においてNP96細胞との差が最も大きいCNE2細胞を選択し、Lipofectamine 2000によるmiR-18a阻害剤、miR-18a阻害剤NC、過剰発現(OE)-TGFBR3またはOE-TGFBR3NCでトランスフェクトしました。 (Invitrogen)。
Lipofectamine 2000(Invitrogen)の仕様に基づいて、M2マクロファージにmiR-18aミミック、miR-18aミミックNC、si-TGFBR3、si-TGFBR3 NC、miR-18a阻害剤、miR-18a阻害剤NC、OE-TGFBR3、またはOE-TGFBR3NC。
M2マクロファージとNPC細胞の共培養
トランスウェルチャンバーでの細胞共培養は、NPC細胞に対するM2マクロファージからのmiRNAの影響を調査するために採用されました。上部チャンバーは、孔径が0.4μmのM2マクロファージで満たされ、上部チャンバーの細胞の通過を阻止するだけで、小胞、成長因子、栄養素などの細胞から分泌される小分子は阻止しませんでした。下部チャンバーNPC細胞で拡散しました。
SUNE-1およびCNE2は通常のFBS(Gibco)で培養されました。対数増殖期の細胞を実験に採用しました。
SUNE-1およびCNE2細胞は、孔径0.4μmのTranswellインサート細胞培養皿(Coring、Corning、NY、USA)で、10%FBS-RPMI-1640培地(両方ともGibco製)でM2マクロファージと共培養しました。 。
SUNE-1細胞は、M2マクロファージと共培養されていないか、M2マクロファージ、miR-18aがトランスフェクトされたM2マクロファージ、miR-18aがNCがトランスフェクトされたM2マクロファージ、si-TGFBR3がトランスフェクトされたM2マクロファージ、またはsiと共培養されていません。 -TGFBR3NCでトランスフェクトされたM2マクロファージ。
CNE2細胞は、M2マクロファージ、またはM2マクロファージ、miR-18a阻害剤をトランスフェクトしたM2マクロファージ、miR-18a阻害剤NCをトランスフェクトしたM2マクロファージ、OE-TGFBR3をトランスフェクトしたM2マクロファージ、またはOE-TGFBR3と共培養しませんでした。 NCトランスフェクトされたM2マクロファージ。
3-(4,5-ジメチルチアゾール-2-イル)-2,5-ジフェニルテトラゾリウムブロミドアッセイ
細胞生存率は、3-(4,5-ジメチルチアゾール-2-イル)-2,5-ジフェニルテトラゾリウムブロミド(MTT)アッセイによってテストされました。これは、MTTをホルマザンに還元するミトコンドリアデヒドロゲナーゼの活性を測定するために適用される比色アッセイでした。
トリプシン処理し、4×10 4 で96ウェルプレートに播種します 細胞/ウェル、細胞はそれぞれ0、12、24、36、および48時間目に培地を奪われ、MTT溶液(500μL、0.5 g / L)が補充されました。 4時間インキュベートし、上清を廃棄し、細胞を200μLのジメチルスルホキシド溶液とともにインキュベートしました。光学密度(OD、490 nm)値は、マイクロプレートリーダー(ELX808IU、BioTek、VT、USA)で測定しました。各グループは6つの並列ウェルで設定されました。
コロニー形成アッセイ
NPC細胞のコロニー形成能力は、細胞集団依存性と細胞クローン増殖を反映したコロニー形成アッセイによってテストされました。
24時間培養し、0.25%トリプシンで剥離し、300個の細胞を各グループに3つの平行なウェルを備えた35mmディッシュに播種しました。培地を3日ごとに交換し、細胞を1週間培養し、5 mLの4%パラホルムアルデヒドで固定しました。その後、クリスタルバイオレット染色液で細胞を染色し、風乾した。皿を裏返し、グリッドの透明なフィルムを重ね、顕微鏡(オリンパス、東京、日本)でコロニーの数(50個以上の細胞)を数えました。
スクラッチテスト
細胞移動はスクラッチテストによってテストされました。細胞をトリプシン処理し、各グループに3つの平行ウェルを備えた6ウェルプレートに播種し、90%コンフルエンスまで培養しました。次に、細胞を2%FBSを含む培地でインキュベートし、100μLのチップで垂直方向の引っかき傷を描きました。細胞の移動距離を測定するために、倒立顕微鏡下で0時間と24時間に細胞の写真を撮りました。
トランスウェルアッセイ
細胞の浸潤と移動は、トランスウェルアッセイによってテストされました。トランスウェルチャンバーの上部チャンバーを事前に浸漬し、無血清RPMI 1640培地で1:100に希釈した100μLのマトリゲル(コアリング)を添加しました。上部チャンバーと下部チャンバーに200μLと600μLの無血清RPMI1640培地を別々に加えました。続いて、下部チャンバーに10%FBSを含む600μLRPMI1640培地を添加し、上部チャンバーに200μLの細胞懸濁液(12.5×10 4 )を添加しました。 細胞/ mL)。 40時間インキュベートした細胞をクリスタルバイオレット染色液で染色し、綿棒で拭いて、顕微鏡下でマトリゲルを通過する細胞を数えました。
フローサイトメトリー
細胞アポトーシスと細胞周期分布はフローサイトメトリーによって決定されました。
細胞周期分布はヨウ化プロピジウム(PI)染色によって評価されました。細胞を4×10 5 で6ウェルプレートに播種しました 細胞/ウェル、および70〜80%のコンフルエンスまで培養。予冷した70%エタノールで一晩固定し、細胞を遠心分離し(上清を捨て)、RNAase(1 g / L、200μL)とTriton X-100(2μL)を加え、PI染色液で染色しました。 30分。その後、細胞周期分布は、各フレーズ(G0 / G1フレーズ、Sフレーズ、およびG2 / Mフレーズ)での異なる細胞蛍光強度に従って、488 nmでフローサイトメーター(BD Bioscience、NJ、USA)によって検出されました。
細胞アポトーシスは、アネキシンV-フルオレセインイソチオシアネート(FITC)とPI二重染色によって測定されました。細胞を500μLの結合バッファーに再懸濁し、5μLのアネキシンV-FITC染色液と10μLのPI溶液で染色しました。細胞アポトーシスはまた、光にさらされることなく30分以内にフローサイトメーター(BDBioscience)によってテストされました。散布図では、左下の象限(Q4)の生細胞はFITC - でした。 / PI − 、右下象限(Q3)の初期のアポトーシス細胞はFITC + でした / PI − 、および右上象限(Q2)の後期の壊死およびアポトーシス細胞はFITC + でした。 / PI + 。アポトーシス率=初期アポトーシス率(Q3)+後期アポトーシス率(Q2)。
ヌードマウスの腫瘍異種移植片
ヌードマウスにおけるNPCのモデルを確立することにより、腫瘍の成長を観察した。対数期の0.25%トリプシンによって分離されたSUNE-1およびCNE2細胞は、5×10 7 の単一細胞懸濁液に構成されました。 細胞/ mL。細胞懸濁液(0.2 mL)をマイクロインジェクターでマウスの右脇の下に注入し、マウスモデルを確立しました。モデル化されたマウスは、特定病原体除去環境で飼育されました。 4日目から開始して、腫瘍の成長を観察し、4日ごとにマウスの体重を測定した。ヌードマウスは注射後20日目に安楽死させ、腫瘍を切除し、電子天秤で体重を測定し、写真を撮りました。
デュアルルシフェラーゼレポーター遺伝子アッセイ
デュアルルシフェラーゼレポーター遺伝子システムを採用して、miR-18aの結合部位とTGFBR3 mRNAの3 '非翻訳領域(UTR)を確認しました。生物学的予測ウェブサイト(http://www.microrna.org/microrna/home.do)を使用して、miR-18aの標的遺伝子を分析し、3 '上のmiR-18aの相補的結合部位の存在を発見しました。 TGFBR3のUTR。デュアルルシフェラーゼレポーター遺伝子アッセイを使用して、TGFBR3がmiR-18aによって直接標的化されたかどうかをさらに検証しました。 TGFBR3 3'UTR結合部位のpmirGLO-TGFBR3-野生型(WT)およびpmirG-LO-TGFBR3-変異型(MUT)を構築した。 TGFBR3-WTまたはTGFBR3-MUTおよびmiR-18aミミックまたはミミックNCを、Lipofectamine 2000(Invitrogen)によってSUNE-1およびCNE2細胞にコトランスフェクトし、48時間インキュベートしました。ルシフェラーゼアッセイキット(Promega、米国ウィスコンシン州マディソン)を使用して細胞を分析しました。
統計分析
分析には、SPSS21.0統計ソフトウェア(IBM Corp. Armonk、NY、USA)を使用しました。データは平均±標準偏差として表されました。 2つのグループ間の違いは t によって分析されました 一元配置分散分析(ANOVA)と、それに続くテューキーの事後検定によって、複数のグループ間の差異を検定します。 P では大きな違いが考慮されました <0.05。
結果
M2マクロファージの識別
アドヒアランス法により濃縮された単球およびM-CSFによって誘導された単球を、健康なドナーの末梢血から収集した。 CD68、CD206、およびCD163の免疫蛍光検出により、in vitroでM-CSFによって誘導されたPBMCが、典型的な分子特性を備えたマクロファージに変化し、要件を満たしていることが確認されました(図1a)。
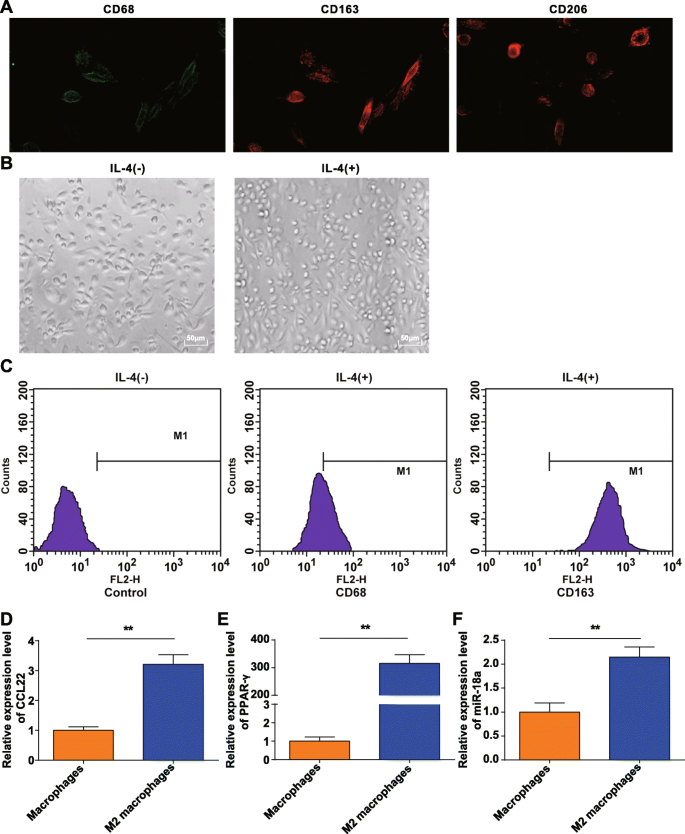
M2マクロファージの同定。 a 。 CD68、CD206、およびCD163は、invitroでの単球誘導後に得られたマクロファージの表面に発現していました。 b IL-4による極性化マクロファージはM2マクロファージでした。 c CD163は高度に発現し、CD68はIL-4によって極性化されたM2マクロファージで低発現しました。 d CCL22はM2マクロファージで高度に発現していました。 e PPAR-γはM2マクロファージで高度に発現していました。 f miR-18aはM2マクロファージで高度に発現していました。 * P <0.05; ** P <0.01。測定データは、平均±標準偏差、 N として表されました。 =3.2つのグループ間の比較は t によって分析されました テスト
得られたマクロファージをIL-4で分極し、顕微鏡で形態を観察した。 IL-4刺激のないマクロファージ(M0)は多様で不規則であり、円形、楕円形、または紡錘形を示していることが明らかになりました。 IL-4によって刺激されると、M2マクロファージは大きくなり、主に丸い形になりました。これは、前述のM2マクロファージの形態学的特徴と一致していました[20](図1b)。
フローサイトメトリーは、20 ng / mLのIL-4で24時間刺激された付着細胞の表面抗原をテストし、CD68が21.16%で発現し、CD163が細胞総数の98.69%であることがわかり(図1c)、付着細胞はM2マクロファージです。 RT-qPCRは、対照的にM0細胞では、CCL22およびPPAR-γ(典型的な分極分子)がM2マクロファージで増加することを明らかにし(図1d、e)、M2マクロファージの誘導が成功したことを示しています。
RT-qPCRは、M0マクロファージとは対照的にM2マクロファージでmiR-18aの発現が増加することも明らかにしました( P ˂0.05)(図1f)。
MiR-18aはNPC細胞で高発現し、TGFBR3は低発現です
バイオインフォマティクスのウェブサイト(miRanda)は、miR-18aの潜在的なターゲットを予測し、TGFBR3はmiR-18aの1つのターゲットと見なされました(図2a)。 miR-18aがTGFBR3の3'UTRを標的としていることを確認するために、デュアルルシフェラーゼレポーター遺伝子アッセイを実施しました。 TGFBR3-WTまたはTGFBR3-MUTをpmirGLOベクターにクローニングし、miR-18aミミックまたはNCでSUNE-1およびCNE2細胞にコトランスフェクトしました。 miR-18a模倣物は、TGFBR3 3'UTR-MUTのルシフェラーゼ活性に影響を与えませんでしたが、SUNE-1およびCNE2細胞におけるTGFBR3 3'UTR-WTのルシフェラーゼ活性を損ない、TGFBR3がmiR-18aによって調節される標的遺伝子であることを示唆しています(図。2b)。
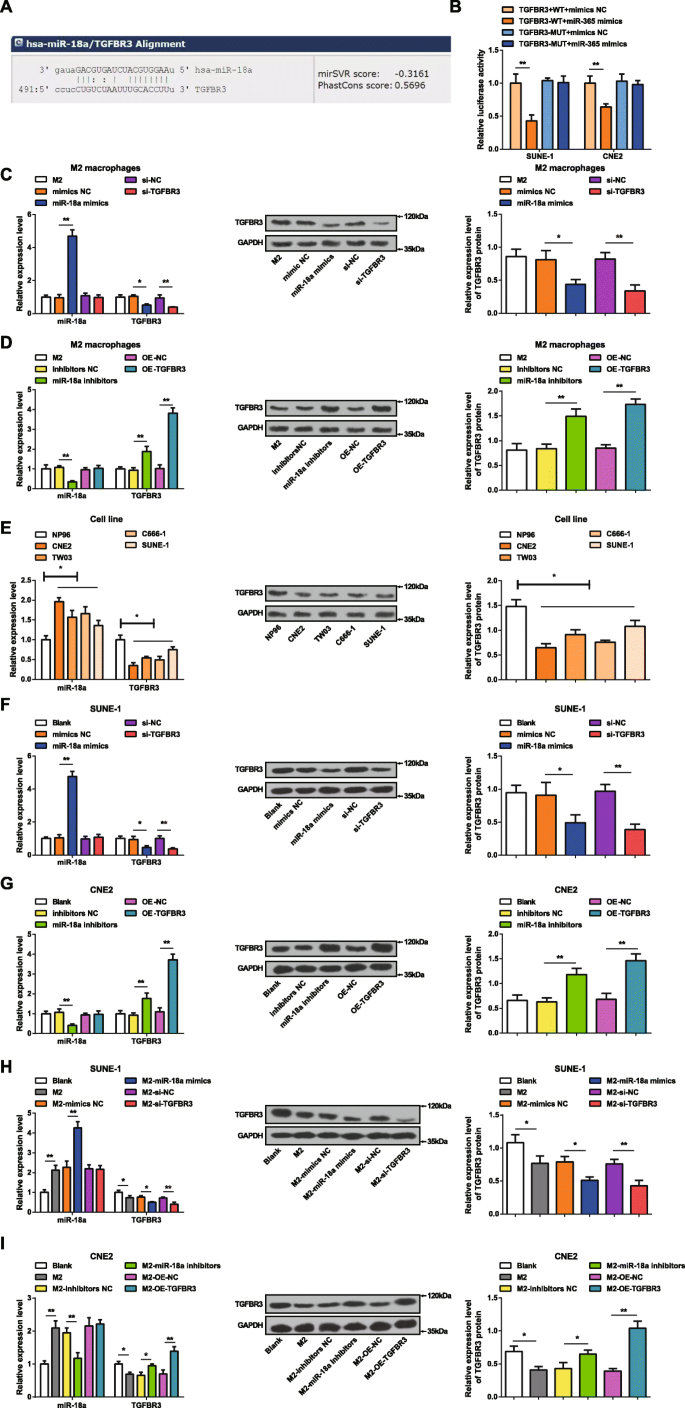
MiR-18aは高度に発現しており、TGFBR3はNPC細胞ではほとんど発現していません。 a miRandaは、TGFBR3をターゲットとするmiR-18aを予測しました。 b デュアルルシフェラーゼレポーター遺伝子アッセイにより、TGFBR3を標的とするmiR-18aが検証されました。 c miR-18aミミックをトランスフェクトしたM2マクロファージでは、miR-18aの発現が上昇し、TGFBR3の発現が低下しました。 d miR-18a阻害剤をトランスフェクトしたM2マクロファージでは、miR-18aの発現が減少し、TGFBR3の発現が上昇しました。 e miR-18aの発現は上昇し、TGFBR3の発現はNP96細胞と比較してNPC細胞株で減少しました。 f miR-18aミミックをトランスフェクトしたSUNE-1細胞では、miR-18aの発現が上昇し、TGFBR3の発現が低下しました。 g miR-18a阻害剤をトランスフェクトしたCNE2細胞では、miR-18aの発現が減少し、TGFBR3の発現が上昇しました。 h miR-18aミミックをトランスフェクトしたM2マクロファージと共培養したSUNE-1細胞では、miR-18aの発現が上昇し、TGFBR3の発現が低下しました。 i miR-18a阻害剤をトランスフェクトしたM2マクロファージと共培養したCNE2細胞では、miR-18aの発現が減少し、TGFBR3の発現が上昇しました。 * P <0.05; ** P <0.01。測定データは、平均±標準偏差、 N として表されました。 =3.2つのグループ間の比較は t によって分析されました テスト。複数のグループ間の比較は、一元配置分散分析とそれに続くテューキーの事後検定によって分析されました
M2マクロファージにおけるmiR-18aミミックまたはmiR-18a阻害剤のトランスフェクション効率は、RT-qPCRおよびウエスタンブロットアッセイによるM2マクロファージにおけるmiR-18aおよびTGFBR3発現の測定によって明らかになりました。 (図2c、d)miR-18aの過剰発現は、M2マクロファージにおけるmiR-18aの発現を上昇させ、TGFBR3の発現を低下させたことは明らかでした。反対に、miR-18a阻害は、M2マクロファージにおけるmiR-18a発現を減少させ、TGFBR発現を増加させました。 M2マクロファージにsi-TGFBR3をトランスフェクトすると、miR-18aの発現に違いは見られませんでしたが、TGFBR3の発現は減少しました。 M2マクロファージへのOE-TGFBR3トランスフェクションでTGFBR3発現が増加した一方で、miR-18a発現に違いは見られませんでした。
CNE2、TW03、C666-1、SUNE-1、およびNP96細胞株におけるmiR-18aおよびTGFBR3の発現は、RT-qPCRおよびウエスタンブロットアッセイによってテストされました。 NPC細胞では、NP96細胞と比較して、miR-18aの発現が増加し、TGFBR3の発現が減少しました(図2e)。 CNE2細胞とSUNE-1細胞は、NP96細胞とのmiR-18a発現の差が最大と最小であるため、それぞれ、進行中のmiRNAダウンレギュレーションとアップレギュレーションアッセイに選択されました。
NPC細胞に対するmiR-18aおよびTGFBR3の効果を特定するために、SUNE-1細胞にmiR-18aミミックまたはsi-TGFBR3をトランスフェクトし、CNE2細胞にmiR-18a阻害剤またはOE-TGFBR3をトランスフェクトしました。 RT-qPCRおよびウエスタンブロットアッセイは、miR-18aがSUNE-1細胞におけるmiR-18a発現の上昇およびTGFBR3発現の低下を模倣することを示しました。 si-TGFBR3のトランスフェクションは、miR-18aの発現に影響を与えませんでしたが、SUNE-1細胞ではTGFBR3の発現が減少しました。 miR-18a阻害剤は、CNE2細胞におけるmiR-18aの発現を減少させ、TGFBR3の発現を上昇させました。 CNE2細胞でのOE-TGFBR3トランスフェクションは、miR-18aの発現には影響しませんでしたが、TGFBR3の発現は上昇しました(図2f、g)。
M2マクロファージからのmiR-18aがNPC細胞に及ぼす影響を調べるために、miR-18aミミックまたはsi-TGFBR3をトランスフェクトしたM2マクロファージまたはmiR-18a阻害剤またはOE-TGFBR3をトランスフェクトしたM2マクロファージをSUNE-1と共培養しました。またはトランスウェルチャンバー内のCNE2細胞。 RT-qPCRおよびウエスタンブロットアッセイでは、SUNE-1またはCNE2細胞におけるmiR-18aおよびTGFBR3の発現をテストしました。トランスフェクトされていないまたはmiR-18aと共培養されたSUNE-1細胞は、トランスフェクトされたM2マクロファージを模倣し、miR-18aの発現が上昇し、TGFBR3の発現が低下することを示しました。 si-TGFBR3をトランスフェクトしたM2マクロファージと共培養したSUNE-1細胞では、miR-18aの発現とTGFBR3の発現の低下に違いは見られませんでした(図2h)。トランスフェクトされていないM2マクロファージとの共培養後、CNE2細胞はmiR-18a発現の増加とTGFBR3発現の減少を特徴としていました。ただし、miR-18a阻害剤をトランスフェクトしたM2マクロファージと事前に共培養したCNE2細胞では、miR-18aの発現が低下しTGFBR3の発現が上昇しました。 miR-18aの発現に違いは認められず、OE-TGFBR3をトランスフェクトしたM2マクロファージと共培養したCNE2細胞ではTGFBR3の発現が増加しました(図2i)。
M2マクロファージのmiR-18aはNPC細胞の生存能力とコロニー形成能力を促進します
MTTアッセイとコロニー形成アッセイを適用して、SUNE-1細胞とCNE2細胞の生存率とコロニー形成能に対するmiR-18aとTGFBR3の影響を特定しました。 miR-18a模倣体、miR-18a阻害剤、si-TGFBR3、またはOE-TGFBR3をSUNE-1細胞またはCNE2細胞にトランスフェクトしました。 SUNE-1細胞では、miR-18aのアップレギュレーションまたはTGFBR3のダウンレギュレーションにより、細胞の生存率が向上し、コロニー数が増加することが示されました(図3a、e)。 CNE2細胞では、miR-18a阻害またはTGFBR3の過剰発現により、細胞の生存率が低下し、コロニー数が減少しました(図3b、f)。
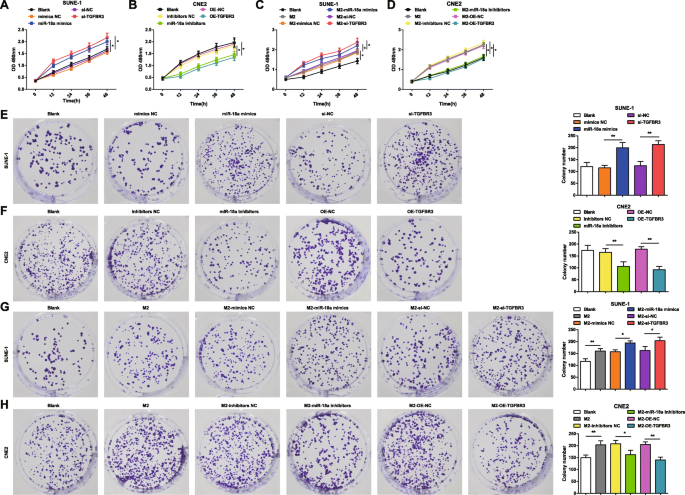
M2マクロファージ由来のmiR-18aは、NPC細胞の生存率とコロニー形成能を誘導します。 a miR-18aミミックまたはsi-TGFBR3はSUNE-1細胞の生存率を増加させました。 b miR-18a阻害剤またはOE-TGFBR3は、CNE2細胞の生存率を低下させました。 c miR-18aミミックまたはsi-TGFBR3をトランスフェクトしたM2マクロファージとの共培養により、SUNE-1細胞の生存率が増加しました。 d miR-18a阻害剤またはOE-TGFBR3をトランスフェクトしたM2マクロファージとの共培養は、CNE2細胞の生存率を低下させました。 e miR-18aミミックまたはsi-TGFBR3は、SUNE-1細胞のコロニー数を増加させました。 f miR-18a阻害剤またはOE-TGFBR3は、CNE2細胞のコロニー数を減少させました。 g miR-18aミミックまたはsi-TGFBR3をトランスフェクトしたM2マクロファージとの共培養により、SUNE-1細胞のコロニー数が増加しました。 h miR-18a阻害剤またはOE-TGFBR3をトランスフェクトしたM2マクロファージとの共培養により、CNE2細胞のコロニー数が減少しました。 * P <0.05; ** P <0.01。測定データは、平均±標準偏差、 N として表されました。 =3.複数のグループ間の比較は、一元配置分散分析とそれに続くテューキーの事後検定によって分析されました
M2マクロファージからのmiR-18aがNPC細胞の生存率とコロニー形成能に及ぼす影響を調べるために、miR-18a模倣体、miR-18a阻害剤、si-TGFBR3-、またはOE-TGFBR3をトランスフェクトしたM2マクロファージを共培養しました。トランスウェルチャンバー内のSUNE-1細胞またはCNE2細胞で。 MTTおよびコロニー形成アッセイは、トランスフェクトされていない、またはmiR-18aミミックでトランスフェクトされたまたはsi-TGFBR3でトランスフェクトされたM2マクロファージと共培養されたSUNE-1細胞が、細胞生存率の強化とコロニー数の増加を示したことを示しました(図3c、g) 。
トランスフェクトされていないM2マクロファージと共培養されたCNE2細胞は、細胞生存率の強化とコロニーの増加で強調されました。しかし、miR-18a阻害剤をトランスフェクトしたまたはOE-TGFBRをトランスフェクトしたM2マクロファージと共培養したCNE2細胞は、細胞生存率の低下とコロニーの減少を示しました(図3d、h)。
M2マクロファージのmiR-18aはNPC細胞の浸潤と移動能力を促進します
miR-18aとTGFBR3がNPC細胞の移動と侵入にどのように影響するかをよりよく理解するために、スクラッチテストとトランスウェルアッセイを実施しました。結果は、miR-18aミミックまたはsi-TGFBR3でトランスフェクトされたSUNE-1細胞が、細胞移動距離と浸潤細胞の増加を特徴としていることを明らかにしました(図4aおよび5a)。
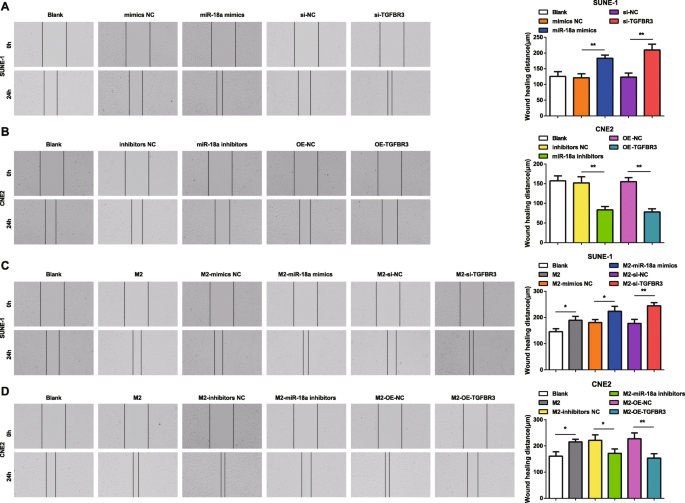
M2マクロファージ由来のmiR-18aは、NPC細胞の遊走能力を促進します。 a miR-18aミミックまたはsi-TGFBR3はSUNE-1細胞の遊走を増加させました。 b miR-18a阻害剤またはOE-TGFBR3は、CNE2細胞の遊走を減少させました。 c miR-18aミミックまたはsi-TGFBR3をトランスフェクトしたM2マクロファージとの共培養により、SUNE-1細胞の遊走が増加しました。 d miR-18a阻害剤またはOE-TGFBR3をトランスフェクトしたM2マクロファージとの共培養は、CNE2細胞の遊走を減少させました。 * P <0.05; ** P <0.01。測定データは、平均±標準偏差、 N として表されました。 =3.複数のグループ間の比較は、一元配置分散分析とそれに続くテューキーの事後検定によって分析されました
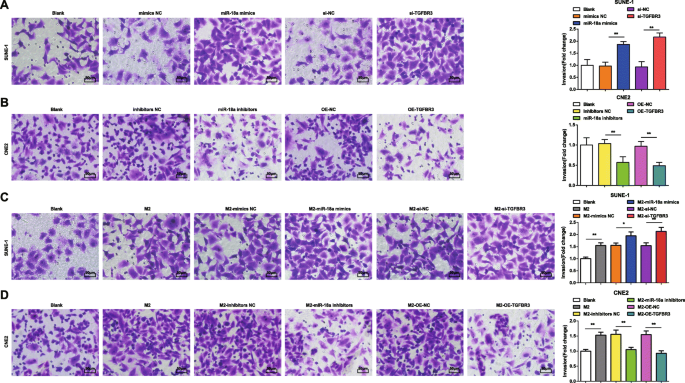
M2マクロファージ由来のmiR-18aは、NPC細胞の浸潤能を促進します。 a miR-18a模倣物またはsi-TGFBR3はSUNE-1細胞の浸潤を増加させました。 b miR-18a阻害剤またはOE-TGFBR3は、CNE2細胞の浸潤を減少させました。 c miR-18aミミックまたはsi-TGFBR3をトランスフェクトしたM2マクロファージとの共培養により、SUNE-1細胞の浸潤が増加しました。 b miR-18a阻害剤またはOE-TGFBR3をトランスフェクトしたM2マクロファージとの共培養により、CNE2細胞の浸潤が減少しました。 * P <0.05; ** P <0.01。測定データは、平均±標準偏差、 N として表されました。 =3.複数のグループ間の比較は、一元配置分散分析とそれに続くテューキーの事後検定によって分析されました
miR-18a阻害剤またはOE-TGFBR3でトランスフェクトされたCNE2細胞では、細胞移動距離と浸潤細胞に減少が見られました(図4bおよび5b)。
M2マクロファージからのmiR-18aがNPC細胞の浸潤および遊走能力にどのように影響したかは、miR-18a模倣体、miR-18a阻害剤、si-TGFBR3-、またはOE-TGFBR3でトランスフェクトされたM2マクロファージとSUNE-1の共培養によって解読されました。またはCNE2細胞。結果は、トランスフェクトされていない、またはmiR-18aミミックトランスフェクトまたはsi-TGFBR3トランスフェクトM2マクロファージと共培養されたSUNE-1細胞が、細胞移動距離および浸潤細胞の増加とともに現れたことを強調しました(図4cおよび5c)。
>トランスフェクトされていないM2マクロファージと共培養されたCNE2細胞では、細胞移動距離と浸潤細胞の両方が増加しました。 miR-18a阻害剤をトランスフェクトしたまたはOE-TGFBR3をトランスフェクトしたM2マクロファージと共培養すると、CNE2細胞は移動距離と浸潤細胞が減少したことが示されました(図4dおよび5d)。
M2マクロファージのmiR-18aは、G0 / G1期に停止するNPC細胞の数を減らし、アポトーシスを抑制します
細胞周期の分布とアポトーシスをフローサイトメトリーでテストし、NPC細胞に対するmiR-18aとTGFBR3の効果を層別化しました。 miR-18aミミックまたはsi-TGFBR3のトランスフェクションにより、G0 / G1期に停止したSUNE-1細胞が減少し、S期およびG2 / M期の細胞が増加し、細胞アポトーシス率が低下したことが示されました(図6aおよび7a)。 。
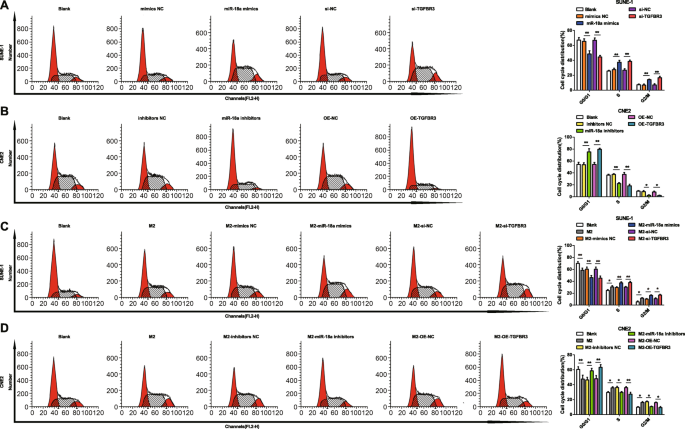
M2マクロファージからのmiR-18aは、G0 / G1期でより少ないNPC細胞を阻止します。 a miR-18aミミックまたはsi-TGFBR3は、G0 / G1期のSUNE-1細胞を減少させ、S期およびG2 / M期のSUNE-1細胞を増加させました。 b miR-18a阻害剤またはOE-TGFBR3は、G0 / G1期のCNE2細胞を増加させ、S期およびG2 / M期のCNE2細胞を減少させました。 c miR-18aミミックまたはsi-TGFBR3でトランスフェクトされたM2マクロファージとの共培養は、G0 / G1期のSUNE-1細胞を減少させ、S期およびG2 / M期のSUNE-1細胞を増加させました。 d miR-18a阻害剤またはOE-TGFBR3でトランスフェクトされたM2マクロファージとの共培養は、G0 / G1期のCNE2細胞を増加させ、S期およびG2 / M期のCNE2細胞を減少させました。 * P <0.05; ** P <0.01。測定データは、平均±標準偏差、 N として表されました。 =3.複数のグループ間の比較は、一元配置分散分析とそれに続くテューキーの事後検定によって分析されました
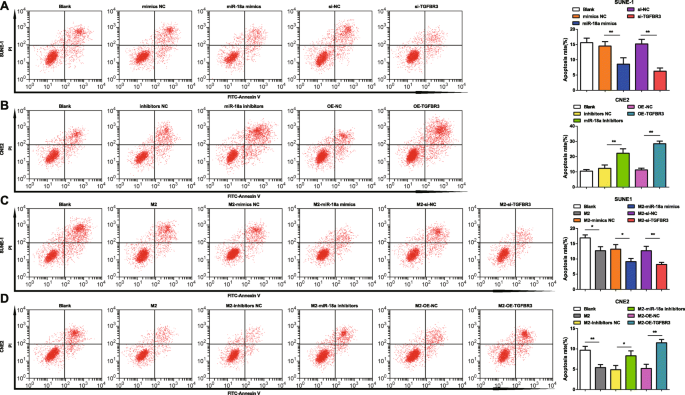
M2マクロファージ由来のmiR-18aはNPC細胞のアポトーシスを阻害します。 a miR-18a模倣物またはsi-TGFBR3はSUNE-1細胞のアポトーシスを減少させました。 b miR-18a阻害剤またはOE-TGFBR3は、CNE2細胞のアポトーシスを増加させました。 c miR-18aミミックまたはsi-TGFBR3をトランスフェクトしたM2マクロファージとの共培養は、SUNE-1細胞のアポトーシスを減少させました。 d miR-18a阻害剤またはOE-TGFBR3をトランスフェクトしたM2マクロファージとの共培養は、CNE2細胞のアポトーシスを増加させました。 * P <0.05; ** P <0.01。測定データは、平均±標準偏差、 N として表されました。 =3.複数のグループ間の比較は、一元配置分散分析とそれに続くテューキーの事後検定によって分析されました
miR-18a阻害剤またはOE-TGFBR3でトランスフェクションすると、G0 / G1期のCNE2細胞は上昇傾向にあり、S期およびG2 / M期のCNE2細胞は低下傾向にあり、細胞アポトーシス率は上昇しました(図6bおよび7b)。 。
NPC細胞周期分布およびアポトーシスにおけるM2マクロファージからのmiR-18aのメカニズムを解読する目的で、miR-18a模倣体、miR-18a阻害剤、si-TGFBR3、またはOE-TGFBR3でトランスフェクトされたM2マクロファージをSUNEと共培養しました。 -トランスウェルチャンバー内の1細胞またはCNE2細胞。トランスフェクトされていない、またはmiR-18aミミックでトランスフェクトされたまたはsi-TGFBR3でトランスフェクトされたM2マクロファージと共培養すると、減少したSUNE-1細胞がG0 / G1期に表示され、増加した細胞がSおよびG2 / M期に表示されました。 1細胞のアポトーシス率が低下しました(図6cおよび7c)。
トランスフェクトされていないM2マクロファージと共培養すると、G0 / G1期のCNE2細胞が減少し、S期とG2 / M期の細胞が増加し、アポトーシス率が低下しました。逆に、miR-18a阻害剤をトランスフェクトまたはOE-TGFBR3をトランスフェクトしたM2マクロファージと共培養すると、G0 / G1期のCNE2細胞が上昇し、S期およびG2 / M期のCNE2細胞が低下し、アポトーシス率が上昇しました(図。6dおよび7d)。
M2マクロファージのmiR-18aはNPCセルのp-Smad1 / t-Smad1を減らし、p-Smad3 / t-Smad3を上げます
ウエスタンブロットアッセイは、NPC細胞でTGFシグナル伝達経路関連タンパク質を検出し、TGFシグナル伝達経路に対するmiR-18aおよびTGFBR3の効果をさらに説明しました。
miR-18aミミックまたはsi-TGFBR3のトランスフェクションにより、SUNE-1細胞のp-Smad1 / t-Smad1が減少し、p-Smad3 / t-Smad3が上昇したことが説明されました(図8a)。
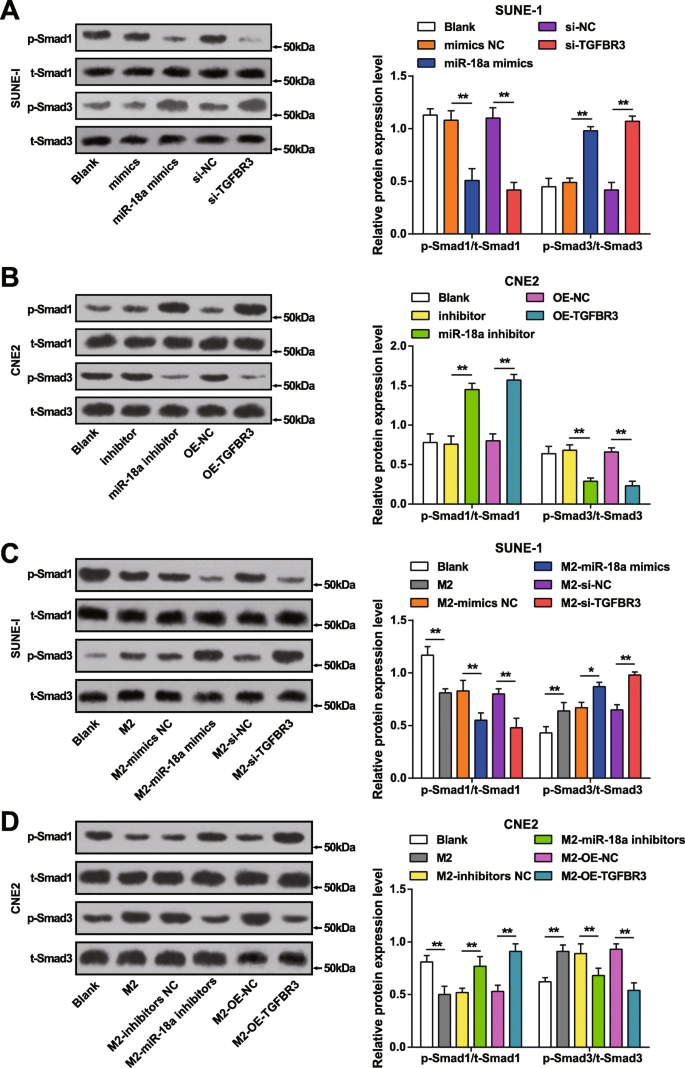
M2マクロファージからのmiR-18aは、NPC細胞でp-Smad1 / t-Smad1を減少させ、p-Smad3 / t-Smad3を増加させます。 a SUNE-1細胞のmiR-18aミミックまたはsi-TGFBR3は、p-Smad1 / t-Smad1を減少させ、p-Smad3 / t-Smad3を上昇させました。 b CNE2細胞におけるmiR-18a阻害剤またはOE-TGFBR3は、p-Smad1 / t-Smad1を増加させ、p-Smad3 / t-Smad3を減少させました。 c miR-18aミミックまたはsi-TGFBR3をトランスフェクトしたM2マクロファージとの共培養により、p-Smad1 / t-Smad1が減少し、p-Smad3 / t-Smad3が上昇しました。 d miR-18a阻害剤またはOE-TGFBR3でトランスフェクトされたM2マクロファージとの共培養は、p-Smad1 / t-Smad1を増加させ、p-Smad3 / t-Smad3を減少させました。 * P <0.05; ** P <0.01。測定データは、平均±標準偏差、 N として表されました。 =3.複数のグループ間の比較は、一元配置分散分析とそれに続くテューキーの事後検定によって分析されました
miR-18a阻害剤またはOE-TGFBR3によるトランスフェクションにより、CNE2細胞でp-Smad3 / t-Smad3が減少し、p-Smad1 / t-Smad1が増加しました(図8b)。
NPC細胞のTGFシグナル伝達経路に影響を与えるM2マクロファージ由来のmiR-18aは、miR-18aミミック-、miR-18aと共培養したSUNE-1細胞およびCNE2細胞のTGFシグナル伝達経路関連タンパク質をテストすることによりウエスタンブロットアッセイによって決定されました。トランスウェルチャンバー内の阻害剤、si-TGFBR3-、またはOE-TGFBR3でトランスフェクトされたM2マクロファージ。
トランスフェクトされていない、miR-18aミミックでトランスフェクトされた、またはsi-TGFBR3でトランスフェクトされたM2マクロファージと共培養されたSUNE-1細胞は、p-Smad1 / t-Smad1の減少とp-Smad3 / t-Smad3の増加で現れました(図8c)。 。
トランスフェクトされていないM2マクロファージと共培養すると、CNE2細胞はp-Smad1 / t-Smad1の低下とp-Smad3 / t-Smad3の上昇に向かう傾向がありました。反対に、CNE2細胞は、miR-18a阻害剤をトランスフェクトしたM2マクロファージまたはOE-TGFBR3をトランスフェクトしたM2マクロファージと共培養すると、p-Smad1 / t-Smad1が増加し、p-Smad3 / t-Smad3が減少することが示されました(図8d)。 。
M2マクロファージのmiR-18aは、NPCを使用したヌードマウスの腫瘍増殖を誘発します
NPCの腫瘍増殖に対するmiR-18aとTGFBR3の影響をさらに解明するために、ヌードマウスに腫瘍異種移植を実施しました。
miR-18aミミックトランスフェクトまたはsi-TGFBR3トランスフェクトSUNE-1細胞を注射すると、マウスは腫瘍体積が拡大し、腫瘍重量が重くなることが示されました(図9a)。
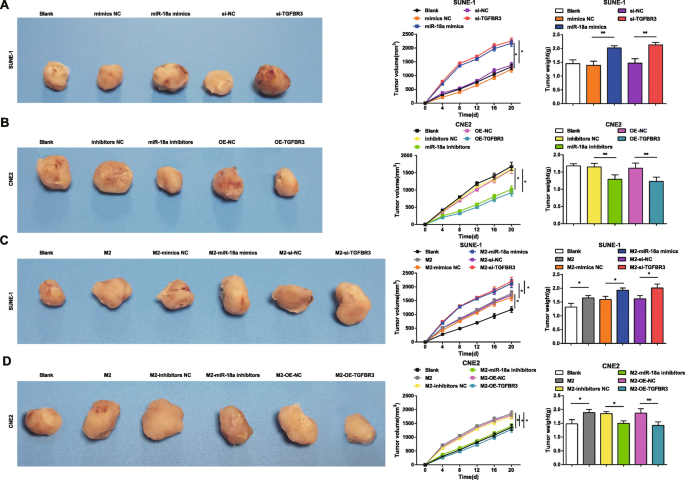
M2マクロファージ由来のmiR-18aは、NPCのヌードマウスの腫瘍増殖を促進します。 a SUNE-1細胞におけるmiR-18a模倣物またはsi-TGFBR3は、腫瘍の体積と重量を増加させました。 b CNE2細胞におけるmiR-18a阻害剤またはOE-TGFBR3は、腫瘍の体積と重量を減少させました。 c miR-18aミミックまたはsi-TGFBR3をトランスフェクトしたM2マクロファージとの共培養により、腫瘍の体積と重量が増加しました。 d miR-18a阻害剤またはOE-TGFBR3をトランスフェクトしたM2マクロファージとの共培養により、腫瘍の体積と重量が減少しました。 * P <0.05; ** P <0.01。測定データは、平均±標準偏差として表され、各グループに3匹のヌードマウスがいた。複数のグループ間の比較は、一元配置分散分析とそれに続くテューキーの事後検定によって分析されました
miR-18a阻害剤をトランスフェクトしたCNE2細胞またはOE-TGFBR3をトランスフェクトしたCNE2細胞を注射したマウスでは、腫瘍の体積と重量の減少が見られました(図9b)。
腫瘍増殖は、miR-18a模倣物、miR-18a阻害剤、si-TGFBR3-、またはOE-TGFBR3をトランスフェクトしたM2マクロファージを注射したマウスで観察され、NPCのM2マクロファージからのmiR-18aのメカニズムを説明しました。
トランスフェクトされていない、miR-18aミミックでトランスフェクトされた、またはsi-TGFBR3でトランスフェクトされたM2マクロファージと共培養した後、SUNE-1細胞をマウスに注入し、マウスの腫瘍体積が大きく、腫瘍重量が重いことが観察されました(図9c)。 P>
CNE2細胞をM2マクロファージと共培養し、マウスに注射した結果、腫瘍の体積と重量が増加していることが示唆されました。 miR-18a阻害剤をトランスフェクトしたまたはOE-TGFBR3をトランスフェクトしたM2マクロファージと共培養したCNE2細胞をマウスに注射すると、腫瘍の体積と重量の両方が減少する傾向がありました(図9d)。
ディスカッション
NPCは、さまざまな要因によって脅かされる多遺伝子性疾患を指します[21]。 miRNAは以前、細胞のプロセスと経路の指標である標的遺伝子の調節を介してNPCの病因に関与することが示唆されていました[22]。具体的には、miR-18aはmiRNA生合成を損なうことでNPCの進行を促進します[6]。この研究は、M2マクロファージからのmiR-18aとNPCにおけるTGFBR3の複合相互作用を解読し、M2マクロファージからのmiR-18aがTGFBR3阻害を介してNPCの進行を刺激するという結論を導き出します(図10)。
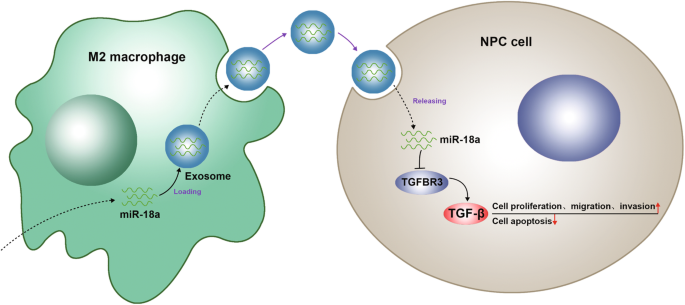
NPCにおけるマクロファージ由来のエクソソームmiR-18aの概略図とTGFBR3を介したTGF-βシグナル伝達経路の関与
この研究の開始時に、マクロファージはIL-4によって刺激され、miR-18aの発現を濃縮することがわかっているM2マクロファージに分化します。ご存知のように、マクロファージの極性化中に、miRNAの発現が変化しました[23]。さらに、M2分極は細胞周期と代謝過程に関与する遺伝子を濃縮し、M2表現型は腫瘍組織の腫瘍成長と血管新生を助長します[24]。 M2マクロファージが豊富なmiR-18aに基づいて、一連の実験が成功裏に実施されました。
当初、私たちの研究では、miR-18aは高度に発現しているのに対し、TGFBR3はNPC細胞ではほとんど発現していないことが明らかになりました。以前の研究から導き出されたように、miR-18aはNPC組織で過剰発現しており、リンパ節転移および臨床病期と関連していると結論付けられています[5]。さらに、miR-18aを含むmiR-17-92クラスターメンバーは、NPC組織で過剰発現していることが報告されています[25]。さらに、アップレギュレーションされたmiR-18aは、腫瘍リンパ節転移の病期と腫瘍サイズに関連するNPC組織で示されることが報告されています[4]。実験的に、舌扁平上皮癌におけるダウンレギュレーションされたTGFBR3 [26]を除いて、望ましくない予後を伴う明細胞腎細胞癌におけるTGFBR3の減少を示す別の研究がありました[27]。とにかく、この研究の結果は、これらの研究結果とある程度一致しています。
NPC細胞の進行におけるmiR-18aとTGFBR3の役割を調査するために、一連のアッセイを実施し、miR-18aの抑制またはTGFBR3の上昇がNPCセルに対する反対の効果。広く、miR-18aの抑制は、卵巣癌、大腸炎関連結腸直腸癌、および肝細胞癌を含む悪性腫瘍における細胞進行を妨げることが証明されています[28、29、30]。狭義には、既存の研究では、アップレギュレーションされたmiR-18aがDICER1の媒介を介してNPC細胞の進行を促進することが明らかになっています[6]。さらに、NPCで過剰発現したmiR-18aはNPC転移と関連していると考えられており、抑制されたmiR-18aはNPC患者の予後の改善に部分的に寄与していることが注目されています[31]。最近、miR-18aのダウンレギュレーションが、NPCの増殖、侵入、および移動を阻止できることが調査されています[4]。さらに、TGFBR3発現の減少は、喉頭扁平上皮癌(LSCC)の浸潤と関連していると見なされ、miR-223 / TGFBR3軸の調節はLSCC進行阻害に関与しています[32]。 TGFBR3の上昇は、NPC細胞の生存を制限し、アポトーシスを誘導し、アポトーシス促進シグナル伝達経路を活性化することが報告されています[14]。以前の研究では、TGFBR3のアップレギュレーションがアポトーシスを促進し、G2 / M期に細胞が停止し、唾液腺腺様嚢胞癌の細胞生存率と遊走が損なわれることが示されています[33]。興味深いことに、TGFBR3の誘導が肝内胆管癌の進行を妨害するのに寄与することが以前に説明されています[34]。
invitroでのNPC細胞における低発現miR-18aおよび過剰発現TGFBR3の保護的役割にもかかわらず、miR-18aノックダウンまたはTGFBR3上昇がヌードマウスの腫瘍増殖を抑制することを説明する結果でさらに検証するために、invivoでヌードミンチで腫瘍異種移植を実施しました。以前の研究で実証されたように、miR-18aを注射したヌードマウスは腫瘍の成長が促進され[5]、逆に、miR-18aアンタゴミールを注射したヌードマウスはNPCで腫瘍の成長が抑制された状態で表示されます[4]。腫瘍増殖におけるTGFBR3の減少に照らして、不十分に発現されたTGFBR3は、淡明細胞型腎細胞癌において腫瘍形成を引き起こすことが示唆されています[27]。反対に、TGFBR3の増加は、トロンボスポンジン1型モチーフ、9アンチセンスRNA 2上昇、およびmiR-223-3p抑制を伴う長鎖ノンコーディングRNA ADAMメタロペプチダーゼの存在により、肺がんの腫瘍増殖を妨げることが認識されています[35 ]。この研究はまた、TGFBR3がmiR-18aの標的遺伝子であることを予測および検証しました。ただし、さらに検証するには、さらに調査を行う必要があります。
結論
一般的に言えば、この研究は、M2マクロファージからのmiR-18aがTGFBR3発現を阻害し、TGF-βシグナル伝達経路を介してNPCの進行を悪化させる具体的なメカニズムを詳しく説明し、その結果はmiR-18aノックダウンまたはTGFBR3上昇によって無効になります。この研究は、NPCの治療目標を更新します。ただし、大規模なコホートの調査は、詳細な調査が必要です。
データと資料の可用性
該当なし。
ナノマテリアル
- ゾル-ゲル法によるナノ構造シリカ/金-セルロース結合アミノ-POSSハイブリッド複合材料とその特性
- PEG化リポソームを介したブファリンの改善された抗腫瘍効果および薬物動態
- パラジウム(II)イオンインプリント高分子ナノスフェアの調製と水溶液からのパラジウム(II)の除去
- 腫瘍の光検出および治療のための5-アミノレブリン酸-スクアレンナノアセンブリ:invitro研究
- 6-メルカプトプリンとニューロン透過性ペプチドで修飾された金ナノ粒子によるSH-SY5Y細胞増殖の促進
- マゲマイトナノ粒子はナノザイムとして機能し、セイヨウアブラナの成長と非生物的ストレス耐性を改善します
- 修飾BiOClの合成と特性評価および水溶液からの低濃度染料の吸着におけるそれらの応用
- 水熱反応によりレモンジュースから製造された蛍光炭素量子ドットの材料と光学特性
- InGaAs / InPコアシェルナノワイヤの自己シードMOCVD成長と劇的に増強されたフォトルミネッセンス
- CpGの改変Fe3O4磁性ナノ粒子送達は、腫瘍増殖および自発的肺転移を阻害して免疫療法を強化する
- 逆ミセル合成によるナノアルギネート:ドキソルビシンカプセル化と乳がん細胞毒性