


hGC33修飾およびソラフェニブ負荷ナノ粒子は、Wntシグナル伝達経路を阻害することにより相乗的な抗肝細胞癌効果を示します
要約
腫瘍特異的阻害剤の送達は、癌治療における課題です。抗体修飾ナノ粒子は、特定の腫瘍関連抗原を過剰発現する腫瘍細胞に、ロードされた薬剤を送達することができます。ここでは、肝細胞癌で過剰発現する膜タンパク質であるグリピカン-3(GPC3 +)に対する抗体hGC33で修飾されたソラフェニブをロードしたポリエチレングリコール-b-PLGAポリマーナノ粒子を構築しました。 hGC33で修飾されたNP(hGC33-SFB-NP)がGPC3 + をターゲットにしていることがわかりました。 肝細胞癌(HCC)細胞は、HCC細胞の表面でGPC3に特異的に結合し、Wnt誘導シグナル伝達を阻害し、サイクリンD1の発現をダウンレギュレートすることでG0 / 1のHCC細胞を阻害し、上皮を阻害することでHCC細胞の遊走を抑制します。間葉移行。 hGC33-SFB-NPは、Ras / Raf / MAPK経路とWnt経路をそれぞれGPC3分子と並行して阻害することにより、HCC細胞の移動、サイクル進行、および増殖を阻害しました。 hGC33-SFB-NPは、in vivoでの肝臓がんの増殖を抑制し、担癌マウスの生存率を改善しました。我々は、hGC33がSFB-NPのHCC細胞へのターゲティングを増加させると結論付けています。 hGC33-SFB-NPは、Wnt経路とRas / Raf / MAPK経路を遮断することにより、HCCの進行を相乗的に阻害します。
はじめに
グリピカン3(GPC3)は、グリセロホスファチジルイノシトールアンカーが関与するメカニズムを介して細胞表面に発現するヘパラン硫酸プロテオグリカンです[1]。 GPC3は発生中にさまざまな組織で発現しますが、その発現はプロモーター領域でのDNAメチル化によってほとんどの成人組織で少なくとも部分的に阻害されます[2]。しかし、GPC3タンパク質は肝細胞癌(HCC)患者の約70%で過剰発現され[3、4]、Wntリガンドとの相互作用を通じて古典的なWntシグナル伝達を刺激し[5]、Wnt /縮れ結合を促進してHCCの成長を促進します[6]。 。古典的なWntシグナル伝達経路の活性化は、HCCの悪性形質転換に関連する最も頻繁なイベントの1つです[7、8]。 Wntシグナル伝達を増加させるGPC3の能力に基づいて、GPC3の過剰発現が古典的なWnt経路を刺激することによってHCCの成長を促進すると仮定しました。
GPC3(524–563)のC末端エピトープを認識する治療用モノクローナル抗体(524–563)(hGC33)は、GPC3(524–563)のC末端エピトープを認識し、HepG2およびHuh-の皮下異種移植片における腫瘍増殖を阻害します。マウスでは7 [9、10]。 hGC33は補完的な決定領域移植によってヒト化されており、その抗癌効果はHepG2異種移植に対してhGC33と同じくらい効果的であり、hGC33は臨床試験で使用されています[11]。これらの結果は、hGC33が重要な抗腫瘍活性を有し、抗GPC3治療が、Wntおよび/または他のシグナル伝達経路を遮断することにより、HCC細胞の増殖および/または生存を直接阻害することを示唆しています。
しかし、HCCの複雑な病因のため、GPC3のみをブロックする効果は限られています[12]。したがって、HCCの病因の詳細なメカニズムを調査し、HCCの診断と予後のための有望なバイオマーカーを特定することは、効果的な治療標的を提供し、患者の予後を改善するのに役立つ可能性があります。抗GPC3抗体は、化学療法剤に対するHCCの感受性を高めるために提案されています[13]。標準的な化学療法薬と組み合わせたhGC33の抗腫瘍活性が最近評価されました[14]。ソラフェニブは、RAF / MEK / ERKによって媒介される細胞シグナル伝達経路を遮断して腫瘍増殖を抑制することにより、腫瘍細胞の増殖を直接阻害できる新規の経口標的HCC薬です[15、16]。ただし、in vivoでのソラフェニブの非標的分布およびHCCでの異常に活性化されたWntシグナルは、薬剤の有効性を制限し、その副作用を増加させます[14、15、16]。 WntはFrizzledファミリーの受容体に結合して、細胞増殖、アポトーシス、細胞遊走を調節する細胞内シグナル伝達を活性化し、HCCなどの多くの腫瘍で薬剤耐性を引き起こします[17、18、19]。
HepG2異種移植モデルでは、hGC33とソラフェニブの組み合わせがソラフェニブ単独よりも腫瘍増殖の抑制に効果的であり[15]、ポリマーナノキャリアによる薬物送達が癌治療で大きな注目を集めています。抗がん剤をロードしたナノキャリアは、in vivoでの非特異的分布と非特異的分解を防ぎ、薬物のバイオアベイラビリティと抗腫瘍ターゲティングを改善し、薬物動態と治療の評価を簡素化できます[15、16]。さまざまなポリマーベースのナノ粒子では、ポリ(乳酸コグリコール酸)(PLGA)ベースの製剤が理想的で安全な薬物担体と見なされています[17、18]。この点で、ポリ(エチレングリコール)- b -ポリ(d、l-ラクチド-co-グリコリド)(PEG- b -PLGA)は、ポリエチレングリコールとPLGAコポリマーに基づいており、加水分解後も安全で毒性がなく、米国食品医薬品局によって承認されています[19、20、21、22]。したがって、PEG- b のナノキャリアの静脈内注射 -PLGA共重合体は、標的化送達を達成し、有効性を高めるための有望な戦略です。さらに、HCC細胞膜上のGPC3分子に対する特異的抗体hGC33の適用は、invitroおよびinvivoでのナノドラッグターゲティングの送達を改善するだけでなく[18、19]、Wntおよび/またはGPC3に関連する他のシグナル経路をブロックし、阻害します癌細胞の増殖および/または生存、および相乗的な抗腫瘍活性を達成する可能性があります。
この研究では、hGC33抗体修飾共重合体PEG- b -PLGAナノ粒子は、invivoおよびinvitroでのHCCへのソラフェニブ(hGC33-SFB-NP)送達の送達を促進し、薬物動態を変化させるHCCアクティブターゲティングを通じてHCC治療の効率を向上させることができます。粒子サイズ、ゼータ電位、粒子形態、薬物捕捉効率、薬物負荷容量、およびin vitro薬物放出に応じて、対象となるNPを包括的に特性評価しました。インビトロターゲティング能力は、HepG2肝癌細胞の細胞取り込みによって特徴付けられます。ソラフェニブとSFB-NPを比較することにより、HCCに対するhGC33-SFB-NPの生体内分布と相乗的治療効果を評価しました。私たちの結果は、hGC33-SFB-NPがGPC3 + をターゲットにできることを示しました HCC。 WntおよびRas / Raf / MAPKシグナル経路を阻害することにより、細胞周期の進行、細胞増殖、および腫瘍浸潤を阻害し、肝臓癌の進行を相乗的に阻害することができます。
材料と方法
資料
hGC33抗体、3-(4,5-ジメチルチアゾール-2-イル)-2,5-ジフェニルテトラゾリウムブロミド(MTT)、4 '、6-ジアミジノ-2-フェニルインドール、二塩酸塩(DAPI)、5,5'、6 、6'-テトラクロロ-1,1 '、3,3'-テトラエチル-ベンズイミダゾリルカルボシアニンヨージド(JC-1)、およびジメチルスルホキシド(DMSO)は、Sigma-Aldrich、Inc。(St。Louis、MO)から入手しました。 1-(3-ジメチルアミノプロピル)-3-エチル-カルボジイミド塩酸塩および N -ヒドロキシスクシンイミドはQiyunBiotech(広州、中国)から入手しました。ビシンコニン酸(BCA)タンパク質定量キット、クマリン-6、およびアネキシンV-FITC / PIアポトーシス検出キットは、Beyotime Biotechnology(上海、中国)から購入しました。 PEG- b -PLGA-マレイミドジブロックコポリマー(mal-PEG- b -PLGA; 25,000〜30,000 Da、PLGA、LA:GA、w / w; PEG、13〜15%)は、Polyscitech(West Lafayette、IN、USA)から購入しました。 PI3K抗体キット(9655#)、p-Akt抗体キット(9916#)、mTOR抗体キット(9964#)、Bcl-2ファミリー抗体キット(9942#)、アポトーシス抗体キット(9915#)、および二次ヤギ抗ウサギおよび抗マウス抗体はCellSignalTechnology(Danvers、MA、USA)から購入しました。サイクリンB1およびサイクリン依存性キナーゼはAbcamBiological Technology(USA)から購入しました。ホスホ-Rb、サイクリンD1、チェックポイントキナーゼ1(CHK1)、P53、リン酸化乳がん感受性遺伝子1(p-BRCA1)、RAD51、チトクロームC、およびマトリックスメタロプロテアーゼ(MMP2およびMMP9)に対する抗体は、Abcam(USA)から購入しました。 。他のすべての分析グレードの化学物質、試薬、および溶媒は、標準的なサプライヤーから入手し、さらに精製することなく使用しました。
細胞と動物
HCC細胞株HepG2(American Type Culture Collection(Manassas、VA、USA)から入手)を、10%FBS(HyClone、Logan、UT、USA)および80を添加したDMEM培地(Invitrogen、Carlsbad、CA、USA)で培養しました。 5%CO 2 の加湿雰囲気でのU / mlペニシリンと80μg/ mlストレプトマイシン 37°Cで。体重20〜22 g(5〜6週間)のBALB / Cヌードマウスは、Nanjing Junke Biotechnology Co.、Ltd。(中国)から提供されました。 BALB / CヌードマウスはSPFルームで飼育されました。すべての動物の世話と治療は、安徽省科学技術大学の動物の世話の要件に従って実施されました。すべての実験プロトコルは、安徽省科学技術大学の動物実験倫理委員会によってレビューおよび承認されています(承認番号:2019dw013)。
NPの準備
NPを準備するには、mal-PEG- b -PLGAとSFBまたはクマリン-6を秤量し、有機相(3:2 v / vジクロロメタン/アセトン)に溶解しました。この溶液を、ポリビニルアルコール(PVA)溶液(5%w / v)に、連続的にボルテックスしながら一滴ずつ加えた。混合物を氷上でプローブソニケーター(出力550 W、8回)で断続的に超音波処理して、油水エマルジョンを作成しました。エマルジョンを、磁気撹拌しながらPVA(1%w / v)溶液に加えた。 SFBとクマリン-6NPは、8000 rpmで30分間遠心分離して収集し、Milli-Q水で3回洗浄しました。
マレイミドと抗体hGC33の遊離スルフヒドリル残基との反応によって形成されるチオエーテル結合によってhGC33-NPを生成するために、hGC33抗体をマレイミド官能化NPと5:1のモル比で混合しました(hGC33:mal-PEG- b -PLGA)、継続的に攪拌しながら4°Cで16時間インキュベートします。 hGC33は、抗体hGC33のスルフヒドリル基とPEG鎖のマレイミド基との反応によりNPに結合しました。非結合抗体は、SepharoseCL-4Bカラムを通過させることにより除去されました。効率的なタンパク質結合は、BCAキット(Thermo Fisher Scientific、米国マサチューセッツ州ウォルサム)で確認されました。
ナノ粒子(NP)の特性評価
形態、粒子サイズ、カプセル化効率(EE)、およびNPの安定性
NPの形態は、透過型電子顕微鏡法(TEM、H-600;日立、東京、日本)で評価された。薬物を含まないhGC33-NPおよび薬物を含まないNPは、臭化カリウムを使用してFTIR分光光度計(Thermo Nicolet、米国ウィスコンシン州マディソン)で記録されました。 NPの平均粒子サイズとゼータ電位は、Malvern Zetasizer ZEN3600 Nano ZS(Malvern Instruments、Malvern、UK)を使用して20°Cで特性評価されました。薬物カプセル化効率(EE)および薬物負荷量(LC)効率は、限外濾過によって評価されました。サンプルを限外ろ過装置(100 kMWCO、ザルトリウス、ゲッティンゲン、ドイツ)にロードし、8000 rpmで25分間、4°Cで遠心分離して遊離薬物を除去しました。同量の各サンプルをアセトニトリルに溶解し、薬物の総量を確認しました。濃度は高速液体クロマトグラフィーで測定した。吸収波長は266nmでした。次の式を使用して、NPの薬物EE(%)を計算しました:(閉じ込められた薬物の重量/薬物の総重量)×100%。 LC(%)は、(カプセル化された薬物の重量/ NPの重量)×100%として計算された。室温でのNPの安定性を理解するために、NPサイズの変化を、所定の時点(0.5、1、2、4、8、12時間、16時間、20時間、および24時間)での動的光散乱(DLS)によって評価しました。 h)25°Cで。
薬物のinvitro薬物放出および細胞取り込みアッセイ
NPからの薬剤は、分子量カットオフが10kDaの透析バッグを使用して調査されました。簡単に説明すると、1 mLのNPを透析バッグ(MWCO 8,000〜10,000 Da、Spectrum Labs Inc.、カリフォルニア州、米国)にロードしました。透析バッグをPBSに浸し、マグネチックスターラーで25°Cで攪拌しました。 NPの薬物放出プロファイルは、100mLの0.2Mリン酸緩衝生理食塩水(PBS; pH =7.4)で7日間測定されました。サンプル中の薬物濃度は、高速液体クロマトグラフィーで測定しました。その後の研究では、hGC33-SFB-NPと同じ粒子サイズのhGC33-クマリン6-NPを使用して、hGC33-SFB-NPのターゲティングを評価しました。 HepG2(GPC3 + )、およびLi-7(GPC3 - )hGC33-クマリン6-NPとともに、5%CO 2、中37°Cで0.5、2、または4時間インキュベートしました。 それぞれ。共培養した細胞を洗浄し、4%ホルムアルデヒドで10分間固定しました。細胞核を5μg/ mLのヘキスト33,342で15分間染色し、細胞内NPの位置を特定しました。細胞内ナノ粒子画像を分析するための共焦点顕微鏡は、共焦点顕微鏡(Olympus FV1000; Olympus Corporation、Tokyo、Japan)で分析されました。
インビトロ細胞効果
遊離hGC33(Ab)、遊離SFB、hGC33-null-NP、またはhGC33-SFB-NPの細胞毒性は、MTTアッセイを使用して決定されました。 HepG2(GPC3 + )セルとLi-7(GPC3 - )対数期の細胞を96ウェルプレートにウェルあたり4000細胞の密度で播種した後、5%CO 2 中37°Cで48時間インキュベートしました。 。細胞をhGC33、遊離SFB、hGC33-null-NP、またはhGC33-SFB-NPで、37°C、5%CO 2 で48時間処理しました。 。一定時間の共培養後、抗細胞増殖活性を記載されているようにMTTアッセイによって決定した[20]。各ウェルの吸光度を490nmで測定し、SPSS 17.0で最大発育阻止濃度値(IC 50)の半分を計算しました。
細胞浸潤能力の測定
対数増殖期の細胞を5×10 4 の密度で6ウェルプレートに播種しました 細胞/ウェル、吸引ヘッドで引っかき、無血清培地と交換。創傷治癒は、対照群と実験群で0時間、24時間、48時間に記録されました。同時に、同じ数の細胞をトランスウェルチャンバーに接種し、遊離のhGC33、遊離のSFB、hGC33-null-NP、またはhGC33-SFB-NPで処理しました。 500マイクロリットルの10%FBS培地を下部チャンバーに加えた。 24時間のインキュベーション後、トランスウェルチャンバーを取り出し、細胞を4%パラホルムアルデヒドで固定し、0.1%クリスタルバイオレットで染色しました。創傷治癒のサイズと遊走細胞の数を計算して、遊走能力を評価しました。
細胞周期の決定
6ウェルプレートで一晩インキュベートした後、細胞を遊離hGC33(Ab)、遊離SFB、hGC33-null-NP、またはhGC33-SFB-NPで24時間処理し、収集してエタノールで固定しました。ヨウ化プロピジウムで染色した後、フローサイトメトリーを実施し、細胞周期をmodifit 3.0(Verity Software House、Topsham、ME)で分析しました。
ウエスタンブロッティング
シグナル経路の活性化状態と標的分子の発現を評価するために、細胞を6ウェルプレートで一晩インキュベートし、遊離hGC33、遊離SFB、hGC33-null-NP、またはhGC33-SFB-NPを24時間適用しました。 。各治療群の細胞を採取し、タンパク質を抽出して測定しました。タンパク質濃度は、BCAタンパク質キット(Biosharp、合肥、中国)を使用して測定および較正されました。サンプルのタンパク質を12個の硫酸アルキルポリアクリルアミドゲル電気泳動で分離し、PVDFメンブレンに転写し、無脂肪ミルクで密封しました。一次抗体(1:1000に希釈)を4℃で一晩インキュベートし、二次抗体(1:2000)を室温で1時間インキュベートしました。バンドはECL基板(Thermo Fisher Scientific Waltham、MA、USA)で視覚化され、画像はゲル画像解析システムで表示され、β-アクチンがコントロールとして使用されました。
InVivo抗腫瘍活動
インビボでのHCCの増殖に対する遊離hGC33、遊離SFB、hGC33-null-NP、SFB-NP、およびhGC33-SFB-NPの阻害を決定した。安徽工業大学の倫理委員会の動物の健康に関する規制とガイドラインに従って、すべての実験は、12時間/ 12時間の光/温度制御室(23±2°C)のケージ内のBALB / cマウスで実施されました。ダークサイクル。 5×10 6 を含む50μlの懸濁液 生きたHepG2細胞を、5週齢の雌BALB / cマウス(20〜22 g)の右腹部に皮下注射しました。腫瘍体積が約50mmに達したとき 3 、マウスをランダムに6つのグループに分けた(各グループに10匹のマウス)。通常の生理食塩水コントロールNS(200μLPBS中の200 mg / kg null NP)、hGC33-null-NP(200μLPBS中のhGC33-null-NP、hgc33 =100μg/ kg /時間に相当)、遊離hGC33(hGC33 in 200μLPBS、100μg/ kg /時間)、遊離SFB(SFB用量:8 mg / kg /時間)、SFB-NP(SFB用量:8 mg / kg /時間)、およびhGC33-SFB-NP( SFB =8 mg / kg / time、hGC33 =100μg/ kg / time)を2日ごとに10回尾静脈から注射しました。マウスの体重と腫瘍サイズを4日ごとに測定した。腫瘍体積の計算式は、体積=0.5× L でした。 × W 2 、ここで、LとWはそれぞれ腫瘍の長さと幅を表します。投与4週間後、動物をジエチルエーテルで麻酔し、腫瘍の大きさと重量を測定した。さらに、腫瘍、心臓、肝臓、腎臓、肺、および脾臓を摘出し、4%パラホルムアルデヒド溶液で固定し、パラフィンに包埋し、切片化し、ヘマトキシリンおよびエオシンで染色して、デジタル顕微鏡で組織学的変化を評価しました。
統計分析
データは平均±標準偏差(SD)として表され、SPSS18.0による分散分析によって評価されました。ペアワイズ統計比較は、両側スチューデントのt検定を使用して実行されました。 P の場合、差異は統計的に有意であると見なされました。 <0.05。
結果
NPの特性評価と薬物放出 インビトロ
NPの直径と表面特性は、in vivoでの細胞取り込み、薬物放出、およびNP分布に影響を与えるため、対応するパラメーターを使用して、調製したNPの特性を評価しました。 SFB-NP、hGC33-null-NP、およびhGC33-SFB-NPポリマーの形態、粒子サイズ、および粒子サイズ分布を図1にまとめています。透過型電子顕微鏡で観察したhGC33-SFB-NPの形態(図。1a)は、ファジーエッジのある剛体核を示しています。これは、hGC33がNPの表面に存在することを示しています。 SFB NP、hGC33-null-NP、およびhGC33-SFB-NPの粒子サイズは100〜150 nmの範囲であり、典型的な単峰性の粒子サイズ分布を持っていました。 hGC33-SFB-NPの平均直径(120.2±10.2 nm)は、hGC33-null-NP、SFB-NP、およびnull NPの平均直径よりもわずかに大きかった(図1a、表1)。 hGC33-SFB-NPおよびhGC33-null-NPの表面には、単一の表面を持つ薄い球状の不明瞭なフィルムが存在し、これは、抗体hGC33がNPの表面に存在することを示しています。 hGC33-SFB-NPおよびhGC33-null-NPのサイズの増加により、hGC33フィルムの存在が確認されました。 hGC33-SFB-NPの合成は 1 で確認されました NP調製前のH-NMR(図1b)。 5.2ppmと1.58ppmのピークは、乳酸の-CH3プロトンに割り当てられました。 4.8 ppmのピークは、グリコール酸の-OCH2-に割り当てられました。 3.6〜3.8 ppmのピークは、PEG繰り返し単位の-CH2CH2O-プロトンに割り当てられました。 PEGの表面化学- b -PLGAおよびAb-PEG- b -PLGA NPは、FTIR分光法によっても研究されました(図2c)。 PEGのスペクトル- b -PLGAポリマー、約1750 cm -1 の強いバンド PLGA鎖のカルボニル基(C =O)のストレッチに由来します。 2880 cm -1 のバンド PEG鎖の-CH基の伸長が原因でした。同時に、2520 cm -1 にピークが現れました。 これは、hGC33抗体の-SH伸縮ピークに起因すると考えられます。 1 H NMRおよびFTIRの結果は、抗体がPEG- b のバックボーンにグラフトされていることを示しています。 -PLGAポリマー。
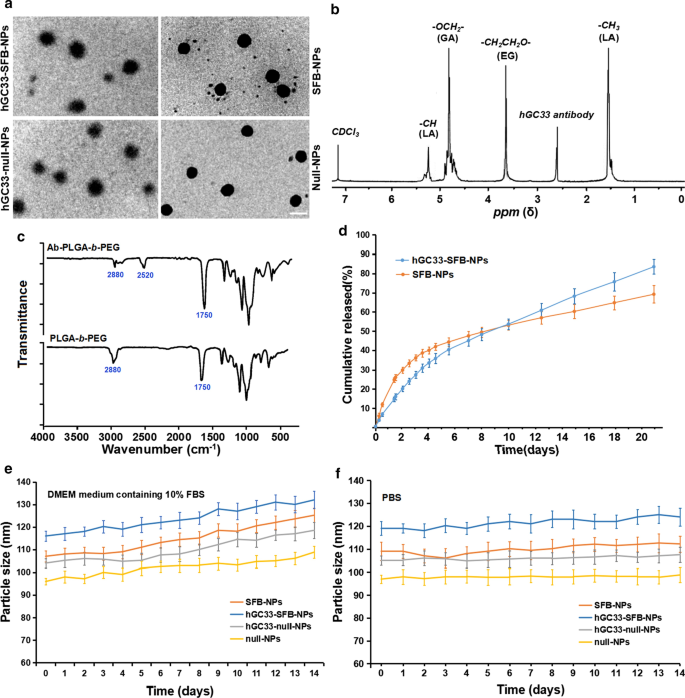
NPの特性。 a NPのTEM特性評価、スケールバーは100nMを示します。 b 1 合成hGC33-PLGA- b のHNMRスペクトル -CDCl3中のPEG; c hGC33-PEG- b のFTIRスペクトル -PLGAおよびPEG- b -PLGA; d 37°CでのPBS(pH =7.4)中のSFB-NPおよびhGC33-SFB-NPの累積放出プロファイル。 e 10%FBSを含むDMEM培地で14日間培養したNPのサイズ変化。 f PBSで14日間インキュベートしたNPのサイズ変化。 SFB、ソラフェニブ; TEM、透過型電子顕微鏡; NP、ナノ粒子; 1 H NMR、 1 H-核磁気共鳴分光法; FTIR、フーリエ変換赤外分光法
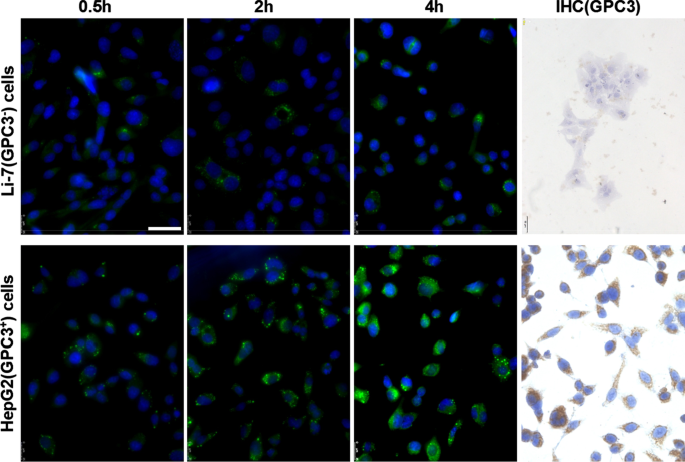
Li-7およびHepG2細胞におけるGPC3の発現とhGC33-クマリン6-NPの取り込み。培養プレートに接種した細胞をPBSで洗浄し、DMEM中の100μg/ mlhGC33-coumarin6-NPと2、4、および8時間インキュベートしました。核をDAPIで染色し、細胞を固定して共焦点レーザー走査顕微鏡で検出しました。 GPC3はLi-7細胞では検出されませんでしたが、免疫細胞化学で検出されたようにHepG2細胞で高度に発現していました。スケールバーは50μMを示します
興味深いことに、10%FBS(pH =7.4)を含むDMEM培地中のhGC33-SFB-NPおよびSFB-NPナノ粒子は、約4日まで急速な薬物放出を示し、その後、比較的ゆっくりと安定した薬物放出を示しました。 20日間にわたるhGC33-SFB-NPとSFB-NPの累積SFB放出は、それぞれ約77%と65%でした(図1d)。違いは、PEG- b の表面の親水性分子に起因する可能性があります -PLGAマトリックス。水和を増加させ、それによって加水分解を促進することにより、ナノ粒子の分解を促進する可能性があります。調製したNPの安定性を測定するために、hGC33-SFB-NP、hGC33-null NP、null-NP、およびSFB-NPを10%FBS(pH =7.4)およびPBS(pH =7.4)を含むDMEM培地に入れました。 )。さまざまなNPのサイズは、2週間以上安定していました。 SFBはhGC33-SFB-NPから14日間持続的かつ安定した方法で放出されましたが、10%FBSの場合と比較して、DMEM培地の粒子のサイズにわずかな変化がありました(図1e、f)。 hGC33-SFB-NPの安定性は、持続的な生物学的役割を持つSFBに適しています。
大きなゼータ電位は、NP間に強い静電反発相互作用を引き起こし、NP分散システムの安定性を維持する可能性があります[23、24]。表1に示すように、hGC33-SFB-NP、hGC33-null-NP、およびSFB-NPのゼータ電位はそれぞれ−18.2±2.2mv、−18.5±1.8mv、および−15.9±2.1mvです。抗体hGC33糖タンパク質のアルデヒド基、カルボキシル基、リン酸基によって生成される負の電荷によって引き起こされます。負に帯電したNPは、NP懸濁液の高い安定性につながる可能性があります。さらに、細胞の表面は通常の生理学的環境では負に帯電しているため、調製されたNPは低電荷で細胞をはじき、組織や細胞への毒性が低くなります。さらに、null-NPおよびSFB-NP(それぞれ多分散度指数[PDI] 0.18および0.19)のサイズ分布は、hGC33-null-NP(PDI =0.21)およびhGC33-SFB-のサイズ分布よりもわずかですが有意に小さくはありませんでした。 NP(PDI =0.23)したがって、ナノ粒子のサイズは良好な均一性を持っています。 NPの粒子サイズ、粒子サイズ分布、およびゼータ電位を、SFBカプセル化効率と負荷量の結果に従って表1に示します。非結合GPC3抗体hGC33を超遠心分離により除去し、NPへのhGC33抗体の結合効率を評価しました。 BCAタンパク質分析は、NPへのhGC33抗体の結合効率が79.5%±2.9%であることを示しました。
hGC33-クマリン6-NPはGPC3を効果的にターゲットにします + HCC細胞株HepG2細胞
ナノ粒子のhGC33抗体がまだGPC3を特異的に標的とする能力を持っているかどうかを調べるために、GPC3 + を使用しました。 HepG2およびGPC3 − 標的細胞としてLi-7細胞、トレーサーナノ粒子としてhGC33-クマリン6-NPを使用し、さまざまな細胞を2時間、4時間、8時間インキュベートしました。細胞をPBSで3回洗浄し、DAPIと反応させて核を染色した。細胞を固定し、ライカ蛍光顕微鏡(DMi8、ドイツ)で写真を撮りました。同じインキュベーション時間で、HepG2細胞の緑色蛍光はLi-7細胞よりも有意に高いことがわかり(図2)、HepG2細胞に入るhGC33-クマリン6-NPの量がそれよりも有意に多かったことを示しています。 Li-7細胞で。結果は、hGC33-Coumarin6-NP上のhGC33抗体が、GPC3を標的とし、ナノ粒子の内在化を仲介する能力を依然として持っていることを示しています。 HepG2細胞およびLi-7細胞におけるGPC3の発現を、間接蛍光および細胞化学的染色で調べました。その結果、HepG2細胞はGPC3を過剰発現し、Li-7細胞はGPC3を発現しなかったことが示されました(図2)。
hGC33-Null-NPはHepG2細胞の増殖を阻害します
hGC33修飾NP(hGC33-null-NP)がHCCの増殖を阻害できるかどうかを判断するために、GPC3陽性HCC細胞株HepG2およびGPC3陰性細胞株Li-7の細胞増殖阻害を測定しました。 hGC33-null-NPとhGC33の両方が24時間の処理後にGPC3陽性HCC細胞株HepG2の増殖を阻害することがわかりましたが、hGC33はhGC33-null-NPよりもHepG2細胞に対してより有意な阻害効果を示しました(図3a);対照的に、hGC33-null-NPおよびhGC33は、GPC3陰性のLi-7細胞株の増殖に影響を与えませんでした(図3b)。 HepG2細胞増殖におけるhGC33-null-NP(0.1μmhgc33に相当)およびhGC33(0.1μm)の代表的な時間経過を図3cに示します。 hGC33はGPC3を認識するC末端ペプチドであるため、hGC33はHepG2の阻害においてhGC33-null-NPよりも優れています。この結果は、hGC33-null-NPのhGC33分子がスペースブロッキング効果を持っている可能性があり、これがhGC33のエピトープへの結合能力に影響を与える可能性があることを示唆しています。
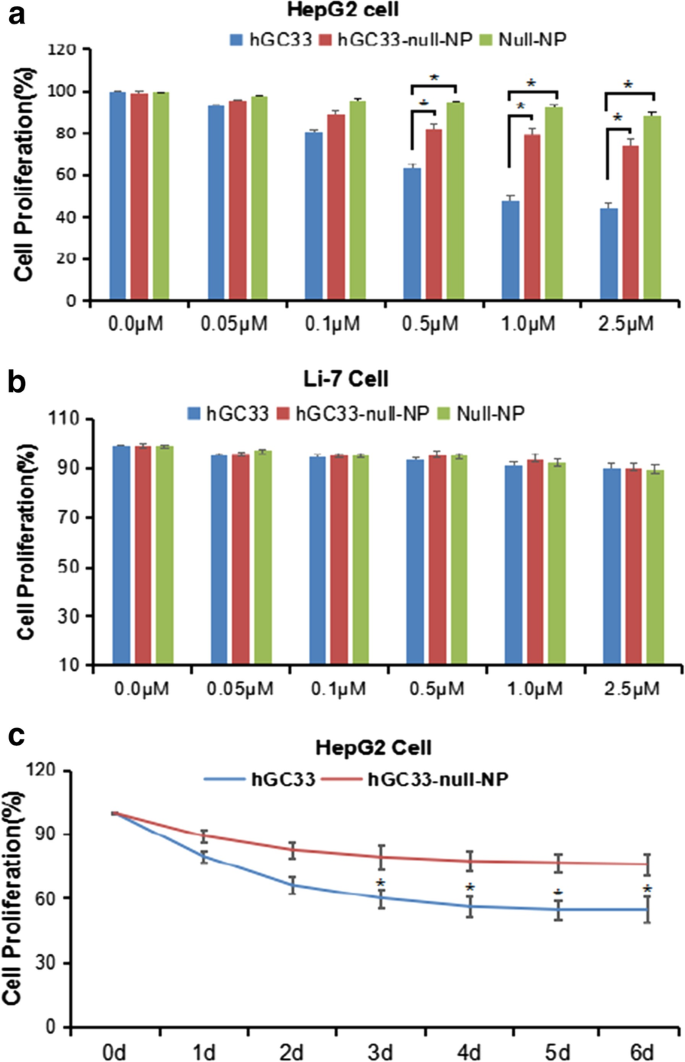
HepG2およびLi-7細胞の増殖は、hGC33、null-NP、およびhGC33-null-NPによって阻害されました。 a 細胞増殖試験は、hGC33、null-NP、またはhGC33-null-NPで処理されたGPC3陽性HepG2細胞で実施されました。 b 細胞増殖試験は、hGC33、null-NP、およびhGC33-null-NPで処理されたGPC3陰性Li-7細胞で実施されました。細胞を0〜2.5μMのhGC33、null-NP、またはhGC33-null-NPと1日間インキュベートしました。細胞の増殖をMTT法で測定し、未処理細胞として標準化した。すべての値は平均±SDを表します。抗体治療なしの対照群(0μM)と比較して、* P <0.01; c hGC33処理(1.0μM)におけるhGC33、null-NP、およびhGC33-null-NPに対するHepG2の応答の代表的な結果
hGC33-Null-NPはHepG2細胞の細胞周期を阻害します
hGC33-null-NPナノ粒子上で修飾されたhGC33抗体の分子活性のメカニズムを理解するために、hGC33-null-NPで処理されたHepG2の細胞周期進行を研究しました。 HepG2細胞株では、hGC33-null-NPとhGC33がG1期の細胞の割合を大幅に増加させ(図4a、b)、hGC33-null-NPとhGC33の両方がG1期の細胞周期停止を誘発する可能性があることを示しています。さらに、hGC33-null-NPおよびhGC33は、HepG2細胞におけるサイクリンD1発現の調節を大幅に低下させる可能性があります(図4c、d)。
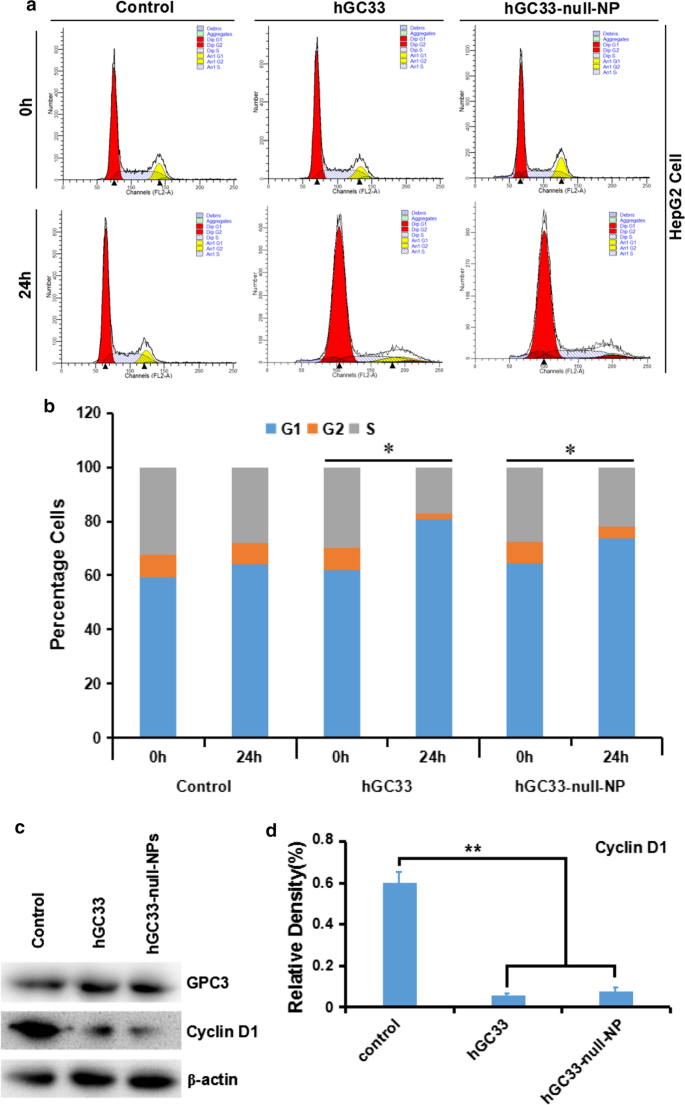
hGC33-null-NPおよびhGC33は、G1期で細胞周期を停止させ、HepG2細胞でのサイクリンD1の発現を抑制しました。 a hGC33-null-NPおよびhGC33で処理されたさまざまなグループの代表的な細胞周期図。 b hGC33-null-NPおよびhGC33で処理されたさまざまなグループの細胞周期分析。 HCC細胞は、0.5μmのhGC33またはhGC33-null-NP(参照としてhGC33のモル濃度)とともにインキュベートされました。 * P <0.05、hGC33-null-NPまたはhGC33細胞のG1期を0時間細胞のG1期と比較しました。 c 、 d hGC33-null-NPまたはhGC33処理後、サイクリンD1は、対照群と比較してHepG2細胞で有意にダウンレギュレーションされました
HepG2、Huh-7、Li-7細胞でのWnt活性化
HCC細胞における古典的なWnt /β-カテニンシグナルの活性化を理解するために、最初に、さまざまなHCC細胞株HepG2(GPC3 ++ )、Huh-7(GPC + )、およびLi-7(GPC3 - )。その結果、β-カテニン経路を伝達するWnt3aおよびFZD受容体は、すべての細胞株、特にHepG2およびHuh-7細胞で発現していることが示されました(図5)。
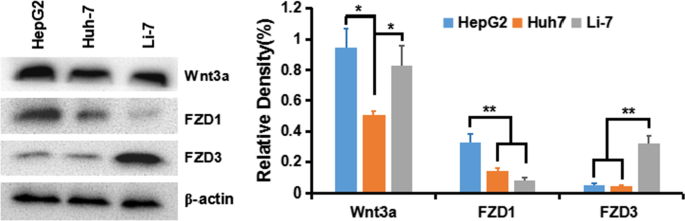
HCC細胞株におけるWnt3a、FZD1、およびFZD3の発現
hGC33-Null-NPは、Wnt3aによって誘導されるWntシグナル伝達に依存する細胞増殖を阻害します
いくつかの研究は、GPC3の細胞外部分がWntの補助受容体である可能性があることを示しています。これはWnt /β-カテニンシグナルの活性化と伝達を促進します。したがって、HepG2(GPC3 + )、Huh-7(GPC3 + )、およびLi-7(GPC3 - )細胞をhGC33およびhGC33-null-NPと共培養し、Wnt /β-カテニンシグナルの活性化をhGC33およびhGC33-null-NPによってブロックし、HepG2およびHuh-7の増殖をブロックしましたが、Li-7はブロックしませんでした。 Wnt3a馴化培地では阻害されました。 Li-7細胞の増殖は、Li-7細胞の表面にGPC3が存在しないことが原因である可能性が最も高いです(図6)。私たちの結果は、hGC33およびhGC33-null-NPナノ粒子が細胞膜上のGPC3分子に特異的に結合することを示しています。 hGC33およびhGC33-null-NPは、Wnt3aのマイトジェン活性を部分的に中和し、Wnt /β-カテニンシグナル伝達経路を阻害します。ただし、hGC33-null-NPナノ粒子による増殖の阻害はhGC33のそれよりも小さかった。おそらく、ナノ粒子の空間構造はhGC33-null-NPを妨害し、ナノ粒子上のhGC33分子の機能を制限するため、GPC3とWnt3aの間の相互作用を完全にブロックすることはできません。
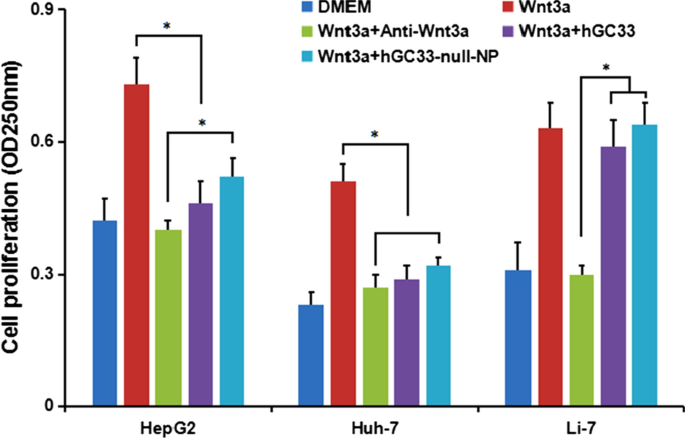
HCCにおけるGPC3活性化Wntシグナル伝達。 50パーセントのWnt3aDMEM培地に、抗wnt3a抗体、hGC33、またはhGC33-null-NPを添加しました。 HepG2(GPC3 ++ )、Huh-7(GPC3 + )およびLi-7(GPC3 − )細胞を48時間共培養し、細胞増殖をMTTアッセイで測定しました。データは平均±SD(* P )として表されました。 <0.01)
hGC33-Null-NPは、HepG2およびHuh-7細胞でWnt3aが誘導するシグナル伝達を阻害します
HCC細胞におけるWnt /β-カテニンシグナル伝達に対するhGC33-null-NPの影響を理解するために、hGC33またはhGC33-null-NPで処理したHepG2およびHuh-7細胞のタンパク質を抽出しました。ウエスタンブロットの結果は、hGC33-null-NP処理後、pYAPおよびpβ-カテニンのレベルは増加したが、YAPおよびβ-カテニンのレベルは減少したことを示した。さらに、hGC33-null-NP群のサイクリンD1、CD44、VEGF、およびc-MYCのレベルは、対照群よりも低かったが、hGC33治療の場合よりも低かった。図7に示すように、HepG2セルとHuh-7セルでも同様の効果が観察されました。
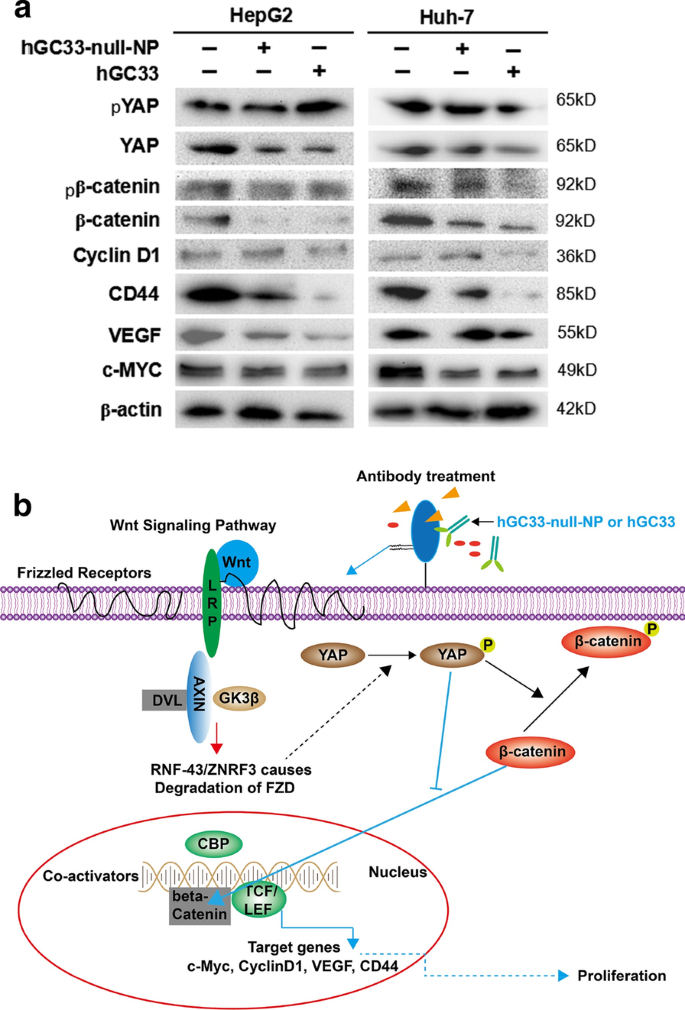
hGC33-null-NPまたはhGC33によるWnt3a誘導性β-カテニンシグナル伝達の阻害。 a 対照群と比較して、hGC33-null-NPまたはhGC33で処理されたHepG2およびHuh-7細胞におけるWnt /β-カテニンシグナル伝達経路は阻害され、β-カテニンおよびYAPのレベルは減少しましたが、リン酸化β-カテニンとリン酸化YAP分子は不安定で、細胞質で分解されました。減少したβ-カテニンは核内で維持することが困難であり、サイクリンD1、CD44、VEGF、およびc-MYCの発現を促進し、その結果、サイクリンD1、CD44、VEGF、およびc-mycタンパク質レベルが減少しました。 b hGC33-null-NPまたはhGC33によって阻害されるWnt /β-カテニンシグナル伝達経路のメカニズムパターン
hGC33-SFB-NPまたはhGC33は、上皮間葉転換(EMT)を阻害することによりHCC細胞の遊走を抑制します
HCCは世界で最も致命的な癌の1つであり、その発生率は着実に増加しています。ソラフェニブは、これらの患者の生存率を改善することが示されているため、進行したHCCの患者に対して承認された唯一の標準治療法です。しかし、臨床的および前臨床的観察は、薬剤耐性の急速な発達のためにソラフェニブ療法の有効性が限られていることを示唆しています。したがって、ソラフェニブに対する逃避抵抗のメカニズムを解明することは、HCC研究の主要な重点です。近年、HCCの進行および薬剤耐性の発生における上皮間葉転換(EMT)の役割にますます注目が集まっています。 EMTとは、上皮から間葉系細胞への形質転換を指し、幹細胞の特徴の獲得、アポトーシスと老化の低減、免疫抑制の促進、癌の発生と発症への関与など、細胞に転移と浸潤の能力を与えます。 E-カドヘリン発現の喪失は、発がんとEMTの重要なステップと考えられています。 EMTは、がん細胞が攻撃性を達成するために使用する、発達上の多段階の分子および細胞の再プログラミングプロセスです。これは主に、E-カドヘリン、ケラチン、ムチン、ZO-1(密着結合タンパク質)の発現をダウンレギュレートすることによるものです。ビメンチン、α-平滑筋アクチン(α-SMA)、FNフィブロネクチン、MMP(分解マトリックス)、N-カドヘリン、カタツムリ、スラッグ、ツイスト、Rho、TGF-β、FGF、I型コラーゲン、およびEMTの特徴的な腫瘍の浸潤、転移、および抗アポトーシスを達成するためのII型コラーゲン。これらのタンパク質発現の変化には、主にWnt /β-カテニンおよびRas / Raf / MAPKシグナル伝達経路の活性化が含まれます。
私たちの実験では、NPベクターの表面にあるhGC33抗体が、Wnt3aによって誘導されるβ-カテニンシグナル伝達を阻害し、β-カテニン、CD44、血管内皮増殖因子(VEGF)、サイクリンD1、およびcの発現をダウンレギュレートできることが示されています。 -私のC。さらに、hGC33-SFB-NPは、Ras / Raf / MAPKシグナル経路の活性化を阻害し、HCC細胞の増殖とアポトーシスを阻害します。 hGC33とSFBには相乗効果があり、EMTを阻害し、HCC細胞の遊走を減少させます(図8)。
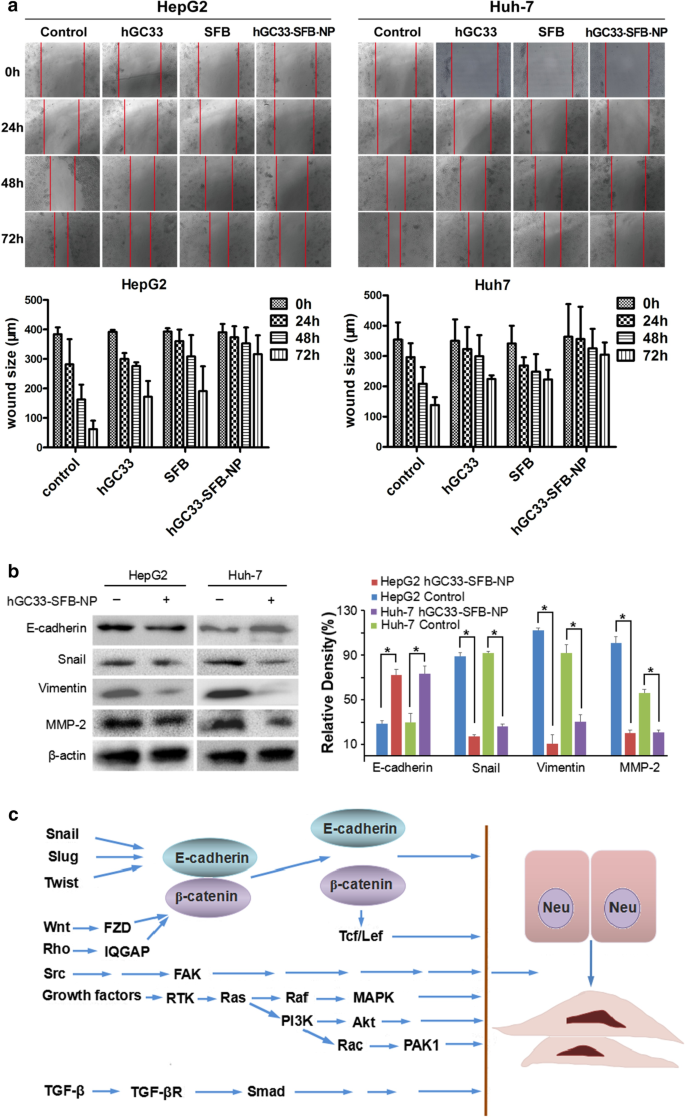
EMT阻害に対するhGC33-SFB-NPの効果。 a 対照群と比較して、hGC33-SFB-NP治療群は細胞移動が少なかった。写真は光学顕微鏡(倍率×200)で撮影した。エラー値は、3回の独立した実験の標準偏差を表します。 *対照群と比較して、 p <0.01。 b 対照群と比較して、hGC33-SFB-NPで処理されたHCCのEMT関連タンパク質カタツムリ、ビメンチン、およびMMP-2は減少しましたが、E-カドヘリンは増加しました。 c EMTの分子メカニズム。 EMT、上皮間葉転換; MMP-2、マトリックスメタロプロテイナーゼ-2; SFB、ソラフェニブ; NP、ナノ粒子
hGC33-SFB-NPは肝細胞癌の増殖を阻害します In Vivo および腫瘍を持ったマウスの生存率を改善
インビボでのhGC33-SFB-NPの抗腫瘍活性を評価するために、HepG2およびHuh-7細胞をそれぞれ雌のBALB / cヌードマウスの右腹部および背側に皮下接種した。腫瘍異種移植片の成長が約30mmに達したとき 3 、マウスをランダムにグループに分けて、各グループ(hGC33-SFB-NP、hGC33-null-NP、SFB-NP、遊離hGC33、遊離SFB、および対照群)の阻害をさらに評価した。HCC効果( n =グループあたり5)。図9a、bから、hGC33-SFB-NPは、PBSコントロールや他の治療法と比較して、マウスの腫瘍増殖を有意に遅らせたことがわかります。 PBSコントロールと比較して、hGC33-null-NP、SFB-NP、遊離hGC33、および遊離SFBもHCCをある程度阻害しました。これは、遊離hGC33および遊離SFBがそれぞれWntシグナルおよびRas / Raf / MAPKを直接阻害するためです。このような経路は、HCC細胞の増殖をある程度阻害する可能性があります。ナノ粒子修飾hGC33(hGC33-null-NP)は化学結合を介してナノ表面に接続されていますが、hGC33によるGPC3分子のターゲティングやWnt活性の阻害には影響しませんでした。ナノ粒子をロードしたSFB(SFB-NP)は、細胞によってエンドサイトーシスされた後、分解されてコポリマーからSFBを放出し、HCCの成長を阻害しました。全体として、HepG2細胞移植片に対するhGC33-SFB-NPの阻害効果は、おそらくHepG2がGPC3分子を発現するため、予想通り、Huh-7細胞移植片よりも大きかった。
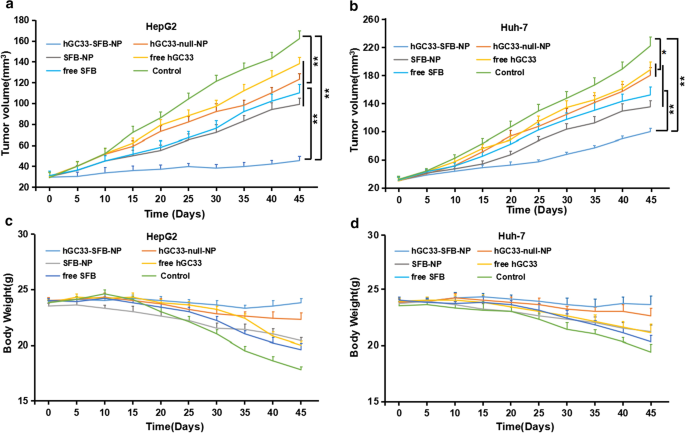
ヌードマウスにおけるHCCの異種移植およびマウス体重の変化に対するhGC33-SFB-NPの効果。各ヌードマウスの背中に肝臓がん細胞を皮下接種した( n =10)。 10日後、担癌マウスをPBS(コントロール)、遊離hGC33、遊離SFB、hGC33-null-NP、SFB-NP、およびhGC33-SFB-NPで処理しました。腫瘍の大きさ( a 、 b )と体重( c 、 d )指定された時点でマウスをモニターしました
図9c、dに示すように、各治療群のヌードマウスの体重も測定しました。対照群の体重は徐々に減少した。遊離hGC33、遊離SFB、SFB-NP、およびhGC33-null-NP治療群のマウスの体重も漸進的に減少し、対照群よりも有意に少なくはありませんでした。しかしながら、hGC33-SFB-NPで治療されたHepG2およびHuh-7を有するヌードマウスの体重はわずかに減少しただけであり、体重は治療サイクルの間比較的安定したままであった。これらの結果は、新規hGC33-SFB-NPナノドラッグがヌードマウスに有意な毒性を持たず、ナノキャリアにロードされたSFBと表面修飾hGC33が相加的または相乗的な抗腫瘍効果を生み出す可能性があることを裏付けています。
ディスカッション
標的HCC療法に対するhGC33-SFB-NPの適合性を調べるために、invitroでHCC細胞上のヒトグリピカン-3に結合する能力についてモデルコンジュゲートをテストしました。グリピカン3陽性HCC細胞の増殖、遊走、およびWnt /β-カテニンシグナル伝達を阻害する。グリピカン3をinvivoで過剰発現するHCCを阻害します。
GPC3特異的抗体hGC33をmal-PEG- b と共有結合させる -PLGAナノ粒子、hGC33のFcセグメントの遊離スルフヒドリル基をマレイミド官能化PEG- b で架橋しました。 -PLGA(mal-PEG- b -PLGA)安定したチオエーテル結合を形成する。接合は、GPC3陽性HCCを標的とするための前提条件です。レンズによって検出されたナノ粒子の直径とゼータ電位の変化、およびhGC33-SFB-NPの細胞内取り込みを含む一連の実験により、hGC33-SFB-NPのHepG2(GPC3 + へのターゲティングが検証されました。 )セル。これらの結果は、抗体hGC33の結合活性がコンジュゲーションによって変化しなかったことを示しています。
GPC3 + の食作用を直接検出しました HepG2およびGPC3 − PEG上のLi-7セル- b -共焦点顕微鏡によるPLGANP表面修飾hGC33。 HepG2およびLi-7細胞をhGC33-クマリン6-NPと共培養した後、HepG2細胞の緑色シグナル強度はLi-7細胞よりも有意に高く、HepG2細胞により多くのナノ粒子が存在することを示しています。この発見は、PEG- b の表面で修飾されたhGC33抗体と一致しています。 -PLGA NPは、HCC細胞の表面でグリピカン3に特異的に結合し、内在化されます。 hGC33で修飾されたNPの内在化の効率は、細胞表面でのGPC3抗原の発現レベルに依存します。
標準的なMTTアッセイを使用して、肝細胞癌細胞の増殖を阻害する効率を測定しました。 hGC33-null-NPとhGC33はどちらもGPC3陽性HCC細胞株HepG2の増殖を阻害しましたが、hGC33-null-NPとhGC33はGPC3陰性Li-7細胞の増殖に影響を与えませんでした(図3b)。動物レベルでは、hGC33-null-NPまたはhGC33のみが、Huh-7およびHepG2異種移植片の成長をある程度阻害しましたが、hGC33-SFB-NPは、マウスでHuh-7およびHepG2肝細胞癌異種移植片の成長停止を引き起こしました。 hGC33-null-NPがGPC3陽性肝細胞癌細胞を有意に阻害したという発見は、PEG- b の阻害効果を示しました。 -HCC細胞増殖におけるPLGANP表面修飾hGC33は、細胞表面でのGPC3抗原の発現に依存します。
GPC3は、WntおよびYAPシグナル伝達を含むHCC病因の多くの経路を調節します[25、26、27]。 GPC3はWntリガンドと相互作用し、Wntの補助受容体であり、HCC増殖のためのWnt / Frizzled結合を促進する可能性があります[28、29]。さらに、肝細胞癌細胞のWntシグナル伝達経路に対するナノドラッグ表面修飾hGC33の効果を調べました。遊離hGC33と同様に、ナノドラッグ表面修飾hGC33は、GPC3を発現するHepG2およびHuh-7細胞におけるWnt誘導シグナル伝達をブロックするだけでなく、Wnt3a誘導β-カテニンおよびYAPシグナル伝達を阻害することによって肝細胞癌細胞の増殖を阻害しました。以前の研究では、YAPの発現はHCCの転写レベルでβ-カテニンによって調節されていることが示されています[30、31]。この研究では、遊離hGC33およびナノドラッグ表面修飾hGC33がWnt3a誘導YAP活性を阻害し、β-カテニン複合体から放出されたYap / TAZも古典的なWntシグナル伝達を開始できることを示しています[32、2]。これらの結果は、典型的なWntとYAPがさまざまなメカニズムを介してクロストークしていることを示しています。 hGC33-null-NPおよびhGC33と比較して、hGC33-SFB-NPは、invitroおよびinvivoでより強力な抗増殖および抗遊走能力を示しました。したがって、hGC33はHCC細胞の特異性を決定するだけでなく、Wnt /β-カテニンやWnt / YAPシグナル伝達など、腫瘍増殖に関連する重要なシグナルをブロックすることにより、HCC細胞の増殖と遊走に対するSFBの阻害効果を高めます。経路。
結論
肝細胞癌細胞で過剰発現する膜タンパク質であるグリピカン-3に対する抗体hGC33は、癌細胞上のグリピカン-3へのソラフェニブ負荷ポリエチレングリコール-b-PLGAポリマーナノ粒子(hGC33-SFB-NP)の結合を増加させました。抗体修飾ナノ粒子の投与は、Wnt誘導シグナル伝達とRas / Raf / MAPKシグナル伝達経路を相乗的に阻害しました。肝細胞癌細胞は、サイクリンD1発現のダウンレギュレーションによって、G0 / 1期に停止し、上皮間葉転換を阻害することによって癌細胞の遊走を抑制しました。 hGC33-SFB-NPは、in vivoでの肝臓がんの増殖を抑制し、担癌マウスの生存率を改善しました。
データと資料の可用性
はい、すべてのデータが原稿に記載されています。
略語
- AKT / PKB:
-
プロテインキナーゼB
- c-MET:
-
HGFR:肝細胞成長因子受容体
- EE%:
-
カプセル化効率%
- EMT:
-
上皮間葉転換
- FTIR:
-
フーリエ変換赤外分光法
- 1 H NMR:
-
1 H核磁気共鳴分光法
- HCC:
-
肝細胞がん
- LC%:
-
薬物負荷%
- MAPK:
-
マイトジェン活性化プロテインキナーゼ
- PDI:
-
多分散度指数
- PI3K:
-
ホスホイノシチド3-キナーゼ
- pRAD51:
-
Phospho-RAD51
- SFB:
-
ソラフェニブ
- TEM:
-
透過型電子顕微鏡
- YAP:
-
はい関連タンパク質-1
ナノマテリアル
- 癌治療のためのナノ粒子:現在の進歩と課題
- コバルトをドープしたFeMn2O4スピネルナノ粒子の調製と磁気特性
- 従来の抗生物質の殺菌効果を活性化するための排出ポンプおよびバイオフィルム阻害剤としてのナノ粒子
- 光触媒活性が強化されたAgナノ粒子/ BiV1-xMoxO4の相乗効果
- ナノ粒子の跳ね返りに及ぼす弾性剛性と表面接着の影響
- 銀ナノ構造の合成方法と応用における最近の進歩
- クルクミンの処理効果を持つPEGコーティングされたCoFe2O4ナノ粒子の毒性
- アナターゼ-ルチル比とTiO2ナノ粒子の光触媒性能に及ぼす酸性解膠剤の影響
- 丸い形の金ナノ粒子:シロイヌナズナの根の成長に対する粒子サイズと濃度の影響
- 金属および金属酸化物ナノ粒子のグリーン合成と単細胞藻類Chlamydomonasreinhardtiiに対するそれらの効果
- ナノ粒子と超音波によって制御される水の過冷却